Gastrointestinal Tract - Abnormalities: Difference between revisions
mNo edit summary |
mNo edit summary |
||
| Line 400: | Line 400: | ||
==Abnormalities of Liver, Biliary Tract, Pancreas, and Spleen== | ==Abnormalities of Liver, Biliary Tract, Pancreas, and Spleen== | ||
For more details on these gastrointestinal tract organs see specific organ pages. | For more details on these gastrointestinal tract organs see specific organ pages. Information below is a summary of {{ICD-11}} information. | ||
{| | {| | ||
| Line 410: | Line 410: | ||
== | ===LB20.0 Structural developmental anomalies of liver=== | ||
[https://icd.who.int/browse11/l-m/en#/http://id.who.int/icd/entity/1737370752 '''LB20''' Structural developmental anomalies of gallbladder, bile ducts or liver] | |||
LB20.00 Fibropolycystic liver disease LB20.21 Biliary atresia | |||
LB20.1 Structural developmental anomalies of gallbladder | |||
===LB20.10 Agenesis, aplasia or hypoplasia of gallbladder=== | |||
===LB20.2 Structural developmental anomalies of bile ducts=== | |||
LB20.20 Choledochal cyst | |||
LB20.21 Biliary atresia | |||
LB20.22 Congenital stenosis or stricture of bile ducts | |||
LB20.23 Structural developmental anomalies of cystic duct | |||
LB20.24 Accessory bile duct | |||
===LB21 Structural developmental anomalies of pancreas=== | |||
[https://icd.who.int/browse11/l-m/en#/http://id.who.int/icd/entity/703933743 '''LB21''' Structural developmental anomalies of pancreas] | |||
LB21.0 Annular pancreas | |||
LB21.1 Pancreas divisum | |||
LB21.2 Accessory pancreas | |||
LB21.3 Agenesis-aplasia of pancreas | |||
LB21.4 Partial agenesis of pancreas | |||
LB21.5 Hypoplasia of pancreas | |||
===LB22 Structural developmental anomalies of spleen=== | |||
[https://icd.who.int/browse11/l-m/en#/http://id.who.int/icd/entity/703933743 '''LB22''' Structural developmental anomalies of spleen] | |||
LB22.0 Congenital asplenia | |||
LB22.1 Polysplenia | |||
LB22.2 Ectopic spleen | |||
==Gastroschisis== | ==Gastroschisis== | ||
Revision as of 10:59, 10 March 2019
| Embryology - 18 Jun 2024 |
|---|
| Google Translate - select your language from the list shown below (this will open a new external page) |
|
العربية | català | 中文 | 中國傳統的 | français | Deutsche | עִברִית | हिंदी | bahasa Indonesia | italiano | 日本語 | 한국어 | မြန်မာ | Pilipino | Polskie | português | ਪੰਜਾਬੀ ਦੇ | Română | русский | Español | Swahili | Svensk | ไทย | Türkçe | اردو | ייִדיש | Tiếng Việt These external translations are automated and may not be accurate. (More? About Translations) |
| ICD-11 |
|---|
|
Structural developmental anomalies of the digestive tract - LB10 Structural developmental anomalies of salivary glands or ducts | LB11 Congenital diverticulum of pharynx | LB12 Structural developmental anomalies of oesophagus | LB13 Structural developmental anomalies of stomach | LB14 Structural developmental anomalies of duodenum | LB15 Structural developmental anomalies of small intestine | LB16 Structural developmental anomalies of large intestine | LB17 Structural developmental anomalies of anal canal | LB18 Congenital anomalies of intestinal fixation Structural developmental anomalies of the liver, biliary tract, pancreas or spleen |
Introduction

The "simple tube" of the gastrointestinal tract and its associated organs have many different tract and organ specific gastrointestinal abnormalities. Due to the complex nature (different germ layer contributions, organogenisis) of the growth, elongation and folding of the tract, there are also several mechanical disorders of folding (rotation). Musculoskeletal abnormalities of the anterior body wall can also result in gastrointestinal abnormalities.
Note that as this system begins function (digestively) postnatally, unless there is a determined genetic history within the family, several abnormalities only become evident postnatally, in particular, metabolic disorders often identified by the Guthrie test.
Some Recent Findings
|
| More recent papers |
|---|
 This table allows an automated computer search of the external PubMed database using the listed "Search term" text link.
More? References | Discussion Page | Journal Searches | 2019 References | 2020 References Search term: Abnormal Development Gastrointestinal Tract <pubmed limit=5>Abnormal Development Gastrointestinal Tract</pubmed> |
Movies
|
|
|
|

Statistics
Australian

|
The pie diagram shows the relative contribution of major gastrointestinal tract abnormalities as a percentage of the total number of congenital abnormalities in Australia beween 1981 - 92.
Note that the digestive system represents approximately 6% of all major congenital abnormalities. One of the most common abnormalities occurring in (2% - 3% population) is Meckel's diverticulum. The mouth (cleft lip, cleft palate) is part of the digestive tract, but more accurately reflects an abnormality of face formation. |
| Data shown as a percentage of all major abnormalities based upon published statistics using the same groupings as Congenital Malformations Australia 1981-1992 P. Lancaster and E. Pedisich ISSN 1321-8352. |
| Australian GIT Abnormalities (2002-2003) |
|---|
Oesophageal atresia/stenosis - (2.0 per 10,000 births) ICD-10 Q39.0–Q39.3
|
Small intestinal atresia/stenosis - (2.4 per 10,000 births) ICD-10 Q41.0-Q41.2
|
Anorectal atresia/stenosis - ( 3.1 per 10,000 births) ICD-10 Q42.0–Q42.3
|
Hirschsprung’s disease - (1.3 per 10,000 births) ICD-10 Q43.1
|
Exomphalos - (Omphalocele) (2.1 per 10,000 births) ICD-10 Q79.2
|
Gastroschisis - (2.6 per 10,000 births) ICD-10 Q79.3
|
|
| Australian Palate Abnormalities (2002-2003) |
|---|
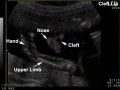
| Cleft lip with or without cleft palate (9.2 per 10,000 births) ICD-10 Q36.0, Q36.1, Q36.9, Q37.0–Q37.5, Q37.8, Q37.9 |
A congenital anomaly characterised by a partial or complete clefting of the upper lip, with or without clefting of the alveolar ridge or the hard palate. Excludes a midline cleft of the upper or lower lip and an oblique facial fissure (going towards the eye).
|
| Cleft palate without cleft lip (8.1 per 10,000 births) ICD-10 Q35.0–Q35.9 |
A congenital anomaly characterised by a closure defect of the hard and/or soft palate behind the foramen incisivum without a cleft lip. This anomaly includes sub-mucous cleft palate, but excludes cleft palate with a cleft lip, a functional short palate and high narrow palate.
|
|
USA Selected
CDC National estimates for selected GIT related major birth defects (2004–2006).
| USA Selected Statistics | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Some Recent Findings
|
| More recent papers |
|---|
This table allows an automated computer search of the external PubMed database using the listed "Search term" text link.
More? References | Discussion Page | Journal Searches | 2019 References | 2020 References Search term: Abnormal Development Gastrointestinal Tract |
Lumen Abnormalities
There are several types of abnormalities (atresia, stenosis and duplication) that impact upon the continuity of the gastrointestinal tract lumen.
Atresia
ICD-11 LB12.1 Atresia of oesophagus | LB14 Structural developmental anomalies of duodenum | LB20.21 Biliary atresia | LB15.1 Atresia of small intestine | LB17.0 Anorectal malformations
An interruption of the lumen where the naming is based upon anatomical location.
Oesophageal Atresia
ICD-11 LB12.1 Atresia of oesophagus
See oesophageal atresia review[7]
| X-ray | Classification[8] |

|

|
The 1962 Waterston classification is a useful tool for predicting post-operative morbidity associated with independent risk factors such as; birth weight, cardiac anomalies, and pre-operative pneumonia.[9] There are alternative prognostic classification systems.[10]
Atresia of oesophagus with tracheo-oesophageal fistula
Atresia of oesophagus with broncho-oesophageal fistula, OA/TOF)
"Oesophageal atresia encompasses a group of congenital anomalies with an interruption in the continuity of the oesophagus, with or without persistent communication with the trachea. In 86% of cases there is a distal tracheooesophageal fistula, in 7% of cases there is no fistulous connection, while in 4% of cases there is a tracheooesophageal fistula without atresia. The remaining cases are made up of patients with OA with proximal, or both proximal and distal, tracheooesophageal fistula."
This abnormality has been shown to be associated with Tbx1 mutations that also include DiGeorge syndrome.[11]
Search PubMed: Oesophageal Atresia
Pyloric Atresia
Pyloric atresia (PA) - a very rare condition (incidence 1 in 100,000 newborns) and about 1% of all intestinal atresias.
Search PubMed: Pyloric Atresia
Duodenal Atresia
ICD-11 LB14 Structural developmental anomalies of duodenum
Search PubMed: Duodenal Atresia
Biliary Atresia
ICD-11 LB20.21 Biliary atresia
Search PubMed: Biliary Atresia
Small Intestine Atresia
ICD-11 LB15.1 Atresia of small intestine
Search PubMed: Small Intestine Atresia
Anorectal Atresia
ICD-11 LB17.0 Anorectal malformations
Search PubMed: Anorectal Atresia
Stenosis
A narrowing of the lumen (esophageal stenosis, duodenal stenosis, pyloric stenosis)
Oesophageal stenosis[12]
Duplication

An incomplete recanalization resulting in parallel lumens, this is really a specialized form of stenosis.
Oesophagus
| ICD-11 LB12 Structural developmental anomalies of oesophagus - Any congenital defect of oesophagus that results from interference with the normal growth and differentiation of the fetus.
LB12.0 Congenital oesophageal web or ring | LB12.1 Atresia of oesophagus | LB12.2 Oesophageal fistula without atresia | LB12.3 Congenital stenosis or stricture of oesophagus | LB12.4 Congenital diverticulum of oesophagus | LB12.5 Congenital dilatation of oesophagus |
LB12.0 Congenital oesophageal web or ring
ICD-11 LB12.0 Congenital oesophageal web or ring - A rare form of incomplete oesophageal obstruction due to a developmental defect of the primitive foregut that presents as a mucosal lesion forming an incomplete diaphragm. Symptoms (apparent from birth) include dysphagia, regurgitation, and choking.
LB12.1 Atresia of oesophagus
ICD-11 LB12.1 Atresia of oesophagus - Oesophageal atresia encompasses a group of congenital anomalies with an interruption in the continuity of the oesophagus, with or without persistent communication with the trachea. In 86% of cases there is a distal tracheooesophageal fistula, in 7% of cases there is no fistulous connection, while in 4% of cases there is a tracheooesophageal fistula without atresia. The remaining cases are made up of patients with OA with proximal, or both proximal and distal, tracheooesophageal fistula.
LB12.2 Oesophageal fistula without atresia
ICD-11 LB12.2 Oesophageal fistula without atresia - This is a birth defect (congenital anomaly) of oesophagus, and one type of EA/TEF, namely isolated "H"-shaped atresia. Tracheoesophageal fistula in which there is no esophageal atresia because the esophagus is continuous to the stomach. Fistula is present between the esophagus and the trachea. Incidence of TE fistula without atresia varies between 1 -11% of esophageal malformations.
LB12.3 Congenital stenosis or stricture of oesophagus
ICD-11 LB12.3 Congenital stenosis or stricture of oesophagus - A form of incomplete oesophageal obstruction due to a developmental defect of the primitive foregut. Abnormal narrowing of the oesophagus occurs most often at the junction of the middle and lower thirds. Clinical manifestations, apparent 2 to 3 weeks after birth, include dysphagia and progressive vomiting.
LB12.4 Congenital diverticulum of oesophagus
ICD-11 LB12.4 Congenital diverticulum of oesophagus - A very rare congenital diverticulum which is typically located just above the cricopharyngeal junction. It is usually asymptomatic unless complicated by an inflammatory process. If the diverticulum compresses the trachea or is associated with oesophageal stenosis or fistula, the symptoms of stridor, progressive dysphagia, respiratory distress, severe choking, and regurgitation may be present from birth.
LB12.5 Congenital dilatation of oesophagus
ICD-11 LB12.5 Congenital dilatation of oesophagus - This is a congenital abnormal enlargement of the lower portion of the esophagus, as seen in patients with achalasia.
Stomach
| ICD-11 LB13 Structural developmental anomalies of stomach - Any congenital defect of stomach that results from interference with the normal growth and differentiation of the fetus.
LB13.0 Congenital hypertrophic pyloric stenosis | LB13.1 Congenital hiatus hernia | LB13.2 Congenital antral web | DA40.2 Gastric volvulus |
LB13.0 Congenital hypertrophic pyloric stenosis
ICD-11 LB13.0 Congenital hypertrophic pyloric stenosis
A not uncommon congenital malformation of the stomach of unknown cause in which there is hypertrophy and hyperplasia of the circular muscle of the pylorus. Symptoms of gastric outlet obstruction usually appear between the third and sixth weeks of life. The anomaly is manifested by intermittent vomiting (which increases in frequency and becomes projectile), regurgitation, weight loss, dehydration, electrolyte imbalance, sometimes a small palpable pyloric mass, and visible peristaltic contractions across the epigastrium; there may also be jaundice. Some cases appear to be familial (possibly of autosomal dominant inheritance).
LB13.1 Congenital hiatus hernia
ICD-11 LB13.1 Congenital hiatus hernia - Congenital diaphragmatic hernia is an embryopathy which is defined by the absence of development of all or part of the diaphragmatic dome that results in the presence of abdominal viscera in the thorax, whit compression of the homolateral lung and impaired development of the controlateral lung.
LB13.2 Congenital antral web
ICD-11 LB13.2 Congenital antral web
DA40.2 Gastric volvulus
ICD-11 DA40.2 Gastric volvulus - Gastric volvulus is an uncommon clinical entity defined as an abnormal rotation (twisting) of all or part of the stomach by more than 180 degrees, creating a closed-loop obstruction of the flow of material through the stomach. It can result in incarceration and strangulation, with variable loss of blood supply. Although rare in childhood, a wandering spleen may also be associated with gastric volvulus, because they share a common etiology: congenital absence of intraperitoneal visceral attachments.
Duodenum
ICD-11 LB14 Structural developmental anomalies of duodenum
Small Intestine
| ICD-11 LB15 Structural developmental anomalies of small intestine - LB15.0 Meckel diverticulum | LB15.1 Atresia of small intestine | LB15.2 Congenital short bowel | LB15.3 Congenital diverticulitis of small intestine | LB15.4 Congenital diverticulosis of small intestine | LB15.5 Congenital diverticulum of small intestine |
LB15.0 Meckel diverticulum
ICD-11 LB15.0 Meckel diverticulum - A congenital abnormality characterized by the outpouching or sac formation in the ILEUM. It is a remnant of the embryonic YOLK SAC in which the VITELLINE DUCT failed to close. During early gestation, the ompahlomesenteric or vitelline duct connects the fetal yolk sac to the primitive gut. By 7-8 weeks of gestation, this duct is normally completely obliterated. A Meckel diverticulum results when this structure fails to resorb completely.



This GIT abnormality is a very common (incidence of 1–2% in the general population) and results from improper closure and absorption of the omphalomesenteric duct (vitelline duct) in development. This transient developmental duct connects the yolk to the primitive gastrointestinal tract.
In addition to Meckel's diverticulum there are a range of other vitelline duct abnormalities, which depend on the degree from a completely patent duct at the umbilicus to lesser remnants (cysts, fibrous cords connecting umbilicus to distal ileum, granulation tissue at umbilicus, or umbilical hernias).
| Historic Embryology | ||
|---|---|---|
|





![97 Short Meckel's Diverticulum]](/embryology/index.php?title=File:Cullen1916_fig97.jpg)






- Links: Yolk Sac - Meckel's Diverticulum | OMIM - Meckel's Diverticulum | Pubmed - Meckel's Diverticulum | Pubmed - omphalomesenteric duct | Pubmed - vitelline duct
LB15.1 Atresia of small intestine
ICD-11 LB15.1 Atresia of small intestine - Jejunoileal atresias and stenoses are major causes of neonatal intestinal obstruction. Atresia refers to a congenital obstruction with complete occlusion of the intestinal lumen. It accounts for 95% of obstructions. Four types of jejunoileal atresias are described. They can range from having a small area of blockage or web to missing large sections of the intestines. Intestinal atresia is one of the most frequent causes of bowel obstruction in the newborn. The ileal atresia is more common than jejunal atresia, and multiple foci are more common than isolated atresia. The most accepted theory regarding the etiology of jejunoileal atresia is that of an intrauterine vascular accident resulting in necrosis of the affected segment. Stenosis, on the other hand, refers to a partial occlusion with incomplete obstruction and accounts for the remaining 5% of cases. A stenosis has an intact mesentery and is a localized narrowing of the bowel. No loss of continuity of the lumen exists.
LB15.2 Congenital short bowel
ICD-11 LB15.2 Congenital short bowel - Short bowel syndrome is a condition in which nutrients are not properly absorbed due to a congenital defect where a large part of the small intestine is missing.
LB15.3 Congenital diverticulitis of small intestine
ICD-11 LB15.3 Congenital diverticulitis of small intestine - This refers to a clinical entity characterized by the presence of sac-like congenital herniations in the wall of the small intestine, in which the pouches of small intestine (diverticula) become infected or inflamed.
LB15.4 Congenital diverticulosis of small intestine
ICD-11 LB15.4 Congenital diverticulosis of small intestine - This refers to a condition characterized by the presence of congenital multiple sack-like mucosal herniations called diverticula through weak points in the wall or lining of the small intestine. Most people with diverticulosis do not have any discomfort or symptoms. However, some people may experience pain or discomfort in the abdomen, bloating, and bleeding.
LB15.5 Congenital diverticulum of small intestine
ICD-11 LB15.5 Congenital diverticulum of small intestine - This refers to a morphological condition in which there is single small congenital pouch in the lining of the small intestine, bulging outward through a weak spot.
Large Intestine
| ICD-11 LB16 Structural developmental anomalies of large intestine - LB16.0 Congenital absence, atresia or stenosis of large intestine | LB16.1 Hirschsprung disease | LB16.2 Immature ganglionosis of large intestine | LB16.3 Congenital hypoganglionosis of large intestine |
LB16.0 Congenital absence, atresia or stenosis of large intestine
ICD-11 LB16.0 Congenital absence, atresia or stenosis of large intestine - Colonic atresia is a congenital intestinal malformation resulting in a non-latent segment of the colon and characterized by lower intestinal obstruction manifesting with abdominal distention and failure to pass meconium in newborns.
LB16.1 Hirschsprung disease
ICD-11 LB16.1 Hirschsprung disease - This is a developmental anomaly affecting the intestinal tract characterized by congenital absence of myenteric ganglion cells (aganglionosis) in a segment of the large bowel. Due to the absence of intrinsic innervation of the muscle layers of the affected segment, there is a loss of motor function. This results in an abnormally large or dilated colon (congenital megacolon) with intestinal occlusion or constipation. This condition becomes evident shortly after birth.
LB16.2 Immature ganglionosis of large intestine
ICD-11 LB16.2 Immature ganglionosis of large intestine - When the number of ganglion cells is normal but the ganglion cells are prominently immature, the disease is referred to as immature ganglionosis or immaturity of ganglia.
LB16.3 Congenital hypoganglionosis of large intestine
ICD-11 LB16.3 Congenital hypoganglionosis of large intestine - The number and size of ganglion cells are small at birth. The size of ganglion cells tends to increase over time, but because their numbers do not increase the symptoms of dysmotility do not improve.Taguchi T, Masumoto K, Ieiri S, Nakatsuji T & Akiyoshi J. (2006). New classification of hypoganglionosis: congenital and acquired hypoganglionosis. J. Pediatr. Surg. , 41, 2046-51. PMID: 17161202 DOI.
Anal Canal
| ICD-11 LB17 Structural developmental anomalies of anal canal - LB17.0 Anorectal malformations | LB17.1 Ectopic anus | LB17.2 Persistent cloaca | LB17.3 Cloacal exstrophy | LB17.4 Perineal groove |
LB17.0 Anorectal malformations
ICD-11 LB17.0 Anorectal malformations - Anorectal malformations (ARMs) are birth defects (due to alterations in embryo development of hindgut or proctodeum) where the anus and rectum (the lower end of the digestive tract) do not develop properly. They occur in approximately 1 in 5000 live births. These comprise a wide spectrum of diseases, which can affect boys and girls, and involve the distal anus and rectum as well as the urinary and genital tracts. Several abnormalities can occur, including the following: A membrane may be present over the anal opening; The rectum may not connect to the anus (imperforate anus); The rectum may connect to a part of the urinary tract or the reproductive system through an abnormal passage called a fistula. The classification of ARMs is mainly based on the position of the rectal pouch relative to the puborectal sling, the presence or absence of fistulas, and the types and locations of the fistulas. The following classification is according to the level of the atretic rectal cul-de-sac with respect to the pubococcygeal line (the radiological landmark for the upper border or the levator ani muscle).
LB17.1 Ectopic anus
ICD-11 LB17.1 Ectopic anus - While children with imperforate or obviously mislocated anus are identified in the newborn period, some children with a very mild abnormality may escape identification until after the newborn period. This mild mislocation of the anus has been termed anterior ectopic anus. Anterior ectopic anus is different from imperforate anus with perineal fistula in that the anal opening is usually of normal size, and only mildly misplaced. Most of these children come to medical attention due to severe constipation.
LB17.2 Persistent cloaca
ICD-11 LB17.2 Persistent cloaca - A congenital anomaly in which the intestinal, urinary, and reproductive ducts open into a common cavity, a result of the failure of the urorectal septum to form during prenatal development. They occur exclusively in girls and comprise the most complex defect in the spectrum of anorectal malformations.
LB17.3 Cloacal exstrophy
ICD-11 LB17.3 Cloacal exstrophy - Rare and complex anorectal and genitourinary malformation in which rectum, vagina and urinary tract share a common everted orifice, accompanied by an omphalocele and an imperforate anus. Exstrophy of the cloaca is a well-known malformation that includes the persistence and the exstrophy of a cloaca that receives ureters, ileum and a rudimentary hindgut. Cloacal exstrophy is a severe birth defect wherein much of the abdominal organs (the bladder and intestines) are exposed. It often causes the splitting of both male and female feitalia (specifically, the penis and clitoris respectively), and the anus is occasionally sealed.
LB17.4 Perineal groove
ICD-11 LB17.4 Perineal groove - The perineal groove describes a normal vestibule but with a groove extending from the vestibule to the anus, which is both normal sized and positioned.
Intestinal Fixation
ICD-11 LB18 Congenital anomalies of intestinal fixation - A condition caused by failure of the intestines to correctly develop during the antenatal period. This condition may present with intermittent abdominal pain, vomiting, or diarrhoea. Confirmation is through observation of intestinal rotation by imaging.
A recent study[14] has suggested that malrotation may result from the stunted embryonic development of intestinal secondary loops.

|
Presents clinically in symptomatic malrotation as:
Neonates - bilious vomiting and bloody stools. Newborn - bilious vomiting and failure to thrive. Infants - recurrent abdominal pain, intestinal obstruction, malabsorption/diarrhea, peritonitis/septic shock, solid food intolerance, common bile duct obstruction, abdominal distention, and failure to thrive. |

Ladd's Bands[15] A series of bands crossing the duodenum which can cause duodenal obstruction. |
Volvulus
ICD-11 DA91.1 Volvulus of small intestine | DB30.1 Volvulus of large intestine
Twisting of the midgut (bowel) which causes obstruction to the flow of material. Can include a variable loss of local blood supply which leads to tissue death.

|

|

|
Diagnosis is generally by upper gastrointestinal radiologic examination or less frequently by barium enema or CT scan.
Corrective surgery is generally by the Ladd's procedure, even with surgical treatment there is still significant associated complications and long-term morbidity.
What abnormal embryological processes could interfere with normal rotation and fixation of the gut?
Search PubMed: intestinal+malrotation
OMIM: Volvulus of Midgut
Links: Medlineplus - childhood volvulus | AAFP - Bilious Vomiting in the Newborn | Pediatric education - Neonatal Bilious Emesis |
Situs Inversus Viscera
Disturbance of the lateralisation of the liver may produce transposition of some or all of the foregut and its derivatives.
Also in situs inversus the anatomical relations of the duodenum, pancreas, bile ducts and portal veins may be reversed or disordered.
Heterotaxia is a term used to describe the rare congenital defect where the major visceral organs are distributed left-right abnormally within the chest and abdomen.
Search PubMed: Situs Inversus Viscera | Heterotaxia
Abnormalities of Liver, Biliary Tract, Pancreas, and Spleen
For more details on these gastrointestinal tract organs see specific organ pages. Information below is a summary of ICD-11 information.
| ICD-11 |
|---|
| LB21 Structural developmental anomalies of pancreas | LB22 Structural developmental anomalies of spleen |
LB20.0 Structural developmental anomalies of liver
LB20 Structural developmental anomalies of gallbladder, bile ducts or liver
LB20.00 Fibropolycystic liver disease LB20.21 Biliary atresia
LB20.1 Structural developmental anomalies of gallbladder
LB20.10 Agenesis, aplasia or hypoplasia of gallbladder
LB20.2 Structural developmental anomalies of bile ducts
LB20.20 Choledochal cyst
LB20.21 Biliary atresia
LB20.22 Congenital stenosis or stricture of bile ducts
LB20.23 Structural developmental anomalies of cystic duct
LB20.24 Accessory bile duct
LB21 Structural developmental anomalies of pancreas
LB21 Structural developmental anomalies of pancreas
LB21.0 Annular pancreas
LB21.1 Pancreas divisum
LB21.2 Accessory pancreas
LB21.3 Agenesis-aplasia of pancreas
LB21.4 Partial agenesis of pancreas
LB21.5 Hypoplasia of pancreas
LB22 Structural developmental anomalies of spleen
LB22 Structural developmental anomalies of spleen
LB22.0 Congenital asplenia
LB22.1 Polysplenia
LB22.2 Ectopic spleen
Gastroschisis
By definition, gastroschisis is a body wall musculoskeletal defect, not a gastrointestinal tract defect, which in turn impacts upon GIT development.
| Gastroschisis (paraomphalocele, laparoschisis, abdominoschisis, abdominal hernia) is an abdominal wall defect associated with evisceration of the intestine (2.5 cases/10,000 births), occuring more frequently in young mothers (less than 20 years old). There have been reports that this abnormality is increasing in incidence.
The abnormality is usually situated to the right of the umbilicus and abdominal contents, mainly gastrointestinal, are found outside the anterior body wall. International Classification of Diseases, 9th Revision diagnosis (756.79) and procedure (54.71) code for gastroschisis (2003 to 2008). |

|
| Can occur in isolation and also in association with other gastrointestinal anomalies (intestinal atresia, perforation, necrosis or volvulus). Defects in other organ systems have been reported in up to 35% of children. Both environments and genetic factors contribute to this disorder. Some studies suggest yolk sac and related vitelline structures[16] or a vascular disruption[17] or are associated with the pathogenesis. A study has also shown that herpesvirus does not play a role in the etiology of gastroschisis.[18]
| |
Gastroschisis patients are commonly small for gestational age (SGA, birth weight < 10th centile). Frequency line graphs of the birth weight distribution.[19]
|

|
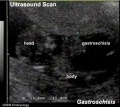
Gastroschisis Ultrasound

|
<html5media height="390" width="440">File:Gastroschisis 001.mp4</html5media> |
Gastroschisis Classification
There has been a recent attempt to classify gastroschisis in order to measure clinical outcomes.[20]
- Simple gastroschisis - intact continuous bowel that is not compromised or breached at delivery or presentation
- Complex gastroschisis - presence of one or more of intestinal atresia, perforation or intestinal necrosis at delivery or presentation, or missed atresia
Omphalocele


The abnormality omphalocele appears similar to gastroschisis, though involves "covered by membranes" and a lack of normal return of the bowel to the abdominal cavity and has a different position relative to the umbilical cord. The abnormality origin differs, as this is a failure of midgut loops to return to the body cavity after initial herniation into the umbilical cord during week 6 - 10.
A recent study in Six4 and Six5 KO mouse has shown similar ventral body wall defects reproducing human omphalocele[1] (More? SIX)
- omphalocele - herniation of the bowel, liver and other organs into the intact umbilical cord, the tissues being covered by membranes unless the latter are ruptured
- gastroschisis - intact umbilical cord and evisceration of the bowel through a defect in the abdominal wall, generally to the right of the cord, with no membrane covering
- Links: Search PubMed - Omphalocele
Short-Bowel Syndrome
ICD-11 KB89.1 Short bowel syndrome
Not generally a developmental abnormality, but related to therapeutic intervention in GIT abnormalities or disease.
Short bowel syndrome is a group of problems affecting people who have had half or more of their small intestine removed. The most common reason for removing part of the small intestine is to treat Crohn's disease. Short bowel syndrome is treated through changes in diet, intravenous feeding, vitamin and mineral supplements, and medicine to relieve symptoms. (NDDIC)
- Links: Better Health Short Bowel syndrome | National Digestive Diseases Information Clearinghouse Short Bowel syndrome
Obstetric Cholestasis
A recent paper in the British Medical Journal discusses this pregnancy associated disease.
"Obstetric cholestasis (or intrahepatic cholestasis of pregnancy) remains widely disregarded as an important clinical problem, with many obstetricians still considering its main symptom, pruritus, a natural association of pregnancy. Obstetric cholestasis is associated with cholesterol gallstones. It may be extremely stressful for the mother but also carries risks for the baby." Piotr Milkiewicz, Elwyn Elias, Catherine Williamson, and Judith Weaver BMJ 2002; 324: 123-124
Small Bowel Obstruction
The are two major forms of small bowel obstruction are from either external (extrinsic) or internal (intrinsic) causes. Listed below are a few examples of both causes.
- Extrinsic Causes - adhesions, closed loop, strangulation, hernia and extrinsic masses
- Intrinsic Causes - intestinal malrotation, Crohn disease, adenocarcinoma, tuberculosis, radiation enteropathy, intramural hemorrhage, intussusception and intraluminal causes.
(More? see PMID 11353110)
Necrotizing Enterocolitis
Necrotizing enterocolitis (NE) is the death of intestinal tissue that occurs postnatally in mainly in premature and low birth weight infants (1 in 2,000 - 4,000 births). The underdeveloped gastointestinal tract appears to be susceptible to bacteria, normally found within the tract,to spread widely to other regions where they damage the tract wall and may enter the bloodstream.
Those with a higher risk for this condition include:
- Premature infants
- Infants who are fed concentrated formulas
- Infants in a nursery where an outbreak has occurred
- Infants who have received blood exchange transfusions
Meconium Plug Syndrome
(functional immaturity of the colon) Term used to describe a transient disorder of the newborn colon, which is characterized by delayed passage of meconium (more than 24 to 48 h), intestinal dilatation and yellow/green vomiting. More common in premature infants and can be determined by radiological dye study.
A recent study[23] by looked at thecorrelation of meconium plug as identified radiologically covering 1994 to 2007, of 77 patients (mean gestational age 37.4 weeks, birth weight, 2977 g) Hirschsprung's disease was found in 10 patients (13%). "Although all patients with plugs and persistent abnormal stooling patterns should prompt a rectal biopsy and genetic probe, the incidence of Hirschsprung's and cystic fibrosis may not be as high as previously reported."
Meconium Ileus
Is a similar meconium obstruction occurring within the small intestine ileum due to abnormal meconium properties possibly associated with abnormalities of small intestine function.
Intestinal Perforation
Usually identified in neonates a disorder due to to one or a combination of:
- necrotizing enterocolitis
- Hirschsprung’s disease
- meconium ileus
Appendix Duplication
Appendix duplication is an extremely rare congenital anomaly (0.004% to 0.009% of appendectomy specimens) first classified according to their anatomic location by Cave in 1936[24] and a later modified by Wallbridge in 1963[25], subsequently two more types of appendix abnormalities have been identified.[26][27]
Modified Cave-Wallbridge Classification (table from[28])
| Classification of types of appendix duplication |
Features |
| A | Single cecum with various degrees of incomplete duplication |
| B1 (bird type) | Two appendixes symmetrically placed on either side of the ileocecal valve |
| B2 (tenia coli type) | ne appendix arises from the cecum at the usual site, and the second
appendix branches from the cecum along the lines of the tenia at various distances from the first |
| B3 | One appendix arises from the usual site, and the second appendix arises from
the hepatic flexura |
| B4 | One appendix arises from the usual site, and the second appendix arises from
the splenic flexura |
| C | Double cecum, each with an appendix |
| Horseshoe appendix | One appendix has two openings into a common cecum |
| Triple appendix | One appendix arises from the cecum at the usual site, and two additional appendixes arise from the colon |
Anorectal Malformations

(ARMs) A group of many different abnormalities that can involve the distal anus and rectum as well as the urinary and genital tracts, for review see[29]. Occurring with an incidence of approximately 1 in 5000 live births.
|
Classification of non-syndromic anorectal malformations (ARM) | |
| Males |
Recto-perineal fistula |
| Recto-urethral-bulbar fistula | |
| Recto-urethral-prostatic fistula | |
| Recto-bladderneck fistula | |
| Imperforated anus without fistula | |
| Complex and unusual defects | |
|
| |
| Females |
Recto-perineal fistula |
| Recto-vestibular fistula | |
| Cloaca with short common channel (< 3 cm) | |
| Cloaca with long common channel (> 3 cm) | |
| Imperforated anus without fistula | |
|
| |
| Complex and unusual defects |
Cloacal extrophy, covered cloacal extra |
| Posterior cloaca | |
| Associated to presacral mass | |
| Rectal atresia | |
|
| |
|
Table Levitt and Peña (2007)[29] | |
Anal Atresia
Anal atresia or imperforate anus is an abnormality of incomplete anorectal region development occurring in about 1 in 5,000 infants. Resulting in accumulation of stool within the colon.
- rectum may end and not connect with the anus
- rectum may connect anatomically to the wrong region (urethra, bladder, or vagina).
- anus may be very narrowed or missing altogether.
Congenital Cloaca
Anal muscles and vagina wall do not form leading to a variable opening composing all or some of the rectum, vagina and bladder. Surgically requires a colostomy and other procedures to transfers a muscle from another part of the body to create a functioning sphincter at the anus.
References
- ↑ 1.0 1.1 Takahashi M, Tamura M, Sato S & Kawakami K. (2018). Mice doubly deficient in Six4 and Six5 show ventral body wall defects reproducing human omphalocele. Dis Model Mech , 11, . PMID: 30237319 DOI.
- ↑ Orgul G, Soyer T, Yurdakok M & Beksac MS. (2018). Evaluation of pre- and postnatally diagnosed gastrointestinal tract obstructions. J. Matern. Fetal. Neonatal. Med. , , 1-6. PMID: 29606013 DOI.
- ↑ Danzer E, Gerdes M, D'Agostino JA, Bernbaum J, Hoffman C, Rintoul NE, Herkert LM, Flake AW, Adzick NS & Hedrick HL. (2015). Patient characteristics are important determinants of neurodevelopmental outcome during infancy in giant omphalocele. Early Hum. Dev. , 91, 187-93. PMID: 25676186 DOI.
- ↑ Pini Prato A, Rossi V, Mosconi M, Holm C, Lantieri F, Griseri P, Ceccherini I, Mavilio D, Jasonni V, Tuo G, Derchi M, Marasini M, Magnano G, Granata C, Ghiggeri G, Priolo E, Sposetti L, Porcu A, Buffa P & Mattioli G. (2013). A prospective observational study of associated anomalies in Hirschsprung's disease. Orphanet J Rare Dis , 8, 184. PMID: 24267509 DOI.
- ↑ Raghoebir L, Biermann K, Buscop-van Kempen M, Wijnen RM, Tibboel D, Smits R & Rottier RJ. (2013). Disturbed balance between SOX2 and CDX2 in human vitelline duct anomalies and intestinal duplications. Virchows Arch. , 462, 515-22. PMID: 23568430 DOI.
- ↑ Ngan ES, Garcia-Barceló MM, Yip BH, Poon HC, Lau ST, Kwok CK, Sat E, Sham MH, Wong KK, Wainwright BJ, Cherny SS, Hui CC, Sham PC, Lui VC & Tam PK. (2011). Hedgehog/Notch-induced premature gliogenesis represents a new disease mechanism for Hirschsprung disease in mice and humans. J. Clin. Invest. , 121, 3467-78. PMID: 21841314 DOI.
- ↑ Spitz L. (2007). Oesophageal atresia. Orphanet J Rare Dis , 2, 24. PMID: 17498283 DOI.
- ↑ Pinheiro PF, Simões e Silva AC & Pereira RM. (2012). Current knowledge on esophageal atresia. World J. Gastroenterol. , 18, 3662-72. PMID: 22851858 DOI.
- ↑ Peters RT, Ragab H, Columb MO, Bruce J, MacKinnon RJ & Craigie RJ. (2017). Mortality and morbidity in oesophageal atresia. Pediatr. Surg. Int. , 33, 989-994. PMID: 28702694 DOI.
- ↑ Yamoto M, Nomura A, Fukumoto K, Takahashi T, Nakaya K, Sekioka A, Yamada Y & Urushihara N. (2018). New prognostic classification and managements in infants with esophageal atresia. Pediatr. Surg. Int. , 34, 1019-1026. PMID: 30099582 DOI.
- ↑ Mc Laughlin D, Murphy P & Puri P. (2014). Altered Tbx1 gene expression is associated with abnormal oesophageal development in the adriamycin mouse model of oesophageal atresia/tracheo-oesophageal fistula. Pediatr. Surg. Int. , 30, 143-9. PMID: 24356861 DOI.
- ↑ Michaud L, Coutenier F, Podevin G, Bonnard A, Becmeur F, Khen-Dunlop N, Auber F, Maurel A, Gelas T, Dassonville M, Borderon C, Dabadie A, Weil D, Piolat C, Breton A, Djeddi D, Morali A, Bastiani F, Lamireau T & Gottrand F. (2013). Characteristics and management of congenital esophageal stenosis: findings from a multicenter study. Orphanet J Rare Dis , 8, 186. PMID: 24289834 DOI.
- ↑ Bower RJ, Sieber WK & Kiesewetter WB. (1978). Alimentary tract duplications in children. Ann. Surg. , 188, 669-74. PMID: 718292
- ↑ Soffers JH, Hikspoors JP, Mekonen HK, Koehler SE & Lamers WH. (2015). The growth pattern of the human intestine and its mesentery. BMC Dev. Biol. , 15, 31. PMID: 26297675 DOI.
- ↑ Panda N, Bansal NK, Narasimhan M & Ardhanari R. (2014). Laparoscopic correction of intestinal malrotation in adult. J Minim Access Surg , 10, 90-2. PMID: 24761085 DOI.
- ↑ Stevenson RE, Rogers RC, Chandler JC, Gauderer MW & Hunter AG. (2009). Escape of the yolk sac: a hypothesis to explain the embryogenesis of gastroschisis. Clin. Genet. , 75, 326-33. PMID: 19419415 DOI.
- ↑ Salinas-Torres VM, Salinas-Torres RA, Cerda-Flores RM & Martínez-de-Villarreal LE. (2018). Genetic variants conferring susceptibility to gastroschisis: a phenomenon restricted to the interaction with the environment?. Pediatr. Surg. Int. , 34, 505-514. PMID: 29550988 DOI.
- ↑ Francis SS, Wiemels JL, Yang W & Shaw GM. (2018). Herpesvirus Infection in Infants with Gastroschisis. Epidemiology , , . PMID: 29634591 DOI.
- ↑ Payne NR, Simonton SC, Olsen S, Arnesen MA & Pfleghaar KM. (2011). Growth restriction in gastroschisis: quantification of its severity and exploration of a placental cause. BMC Pediatr , 11, 90. PMID: 22004141 DOI.
- ↑ Molik KA, Gingalewski CA, West KW, Rescorla FJ, Scherer LR, Engum SA & Grosfeld JL. (2001). Gastroschisis: a plea for risk categorization. J. Pediatr. Surg. , 36, 51-5. PMID: 11150437 DOI.
- ↑ Akakpo-Numado GK, Gnassingbe K, Boume MA, Sakiye KA, Mihluedo-Agbolan K, Attipou K & Tekou H. (2012). Emergency treatment of a ruptured huge omphalocele by simple suture of its membrane. Ann Surg Innov Res , 6, 2. PMID: 22325297 DOI.
- ↑ Matsumaru D, Haraguchi R, Miyagawa S, Motoyama J, Nakagata N, Meijlink F & Yamada G. (2011). Genetic analysis of Hedgehog signaling in ventral body wall development and the onset of omphalocele formation. PLoS ONE , 6, e16260. PMID: 21283718 DOI.
- ↑ Keckler SJ, St Peter SD, Spilde TL, Tsao K, Ostlie DJ, Holcomb GW & Snyder CL. (2008). Current significance of meconium plug syndrome. J. Pediatr. Surg. , 43, 896-8. PMID: 18485962 DOI.
- ↑ Cave AJ. (1936). Appendix Vermiformis Duplex. J. Anat. , 70, 283-92. PMID: 17104589
- ↑ WALLBRIDGE PH. (1962). Double appendix. Br J Surg , 50, 346-7. PMID: 13998581
- ↑ Mesko TW, Lugo R & Breitholtz T. (1989). Horseshoe anomaly of the appendix: a previously undescribed entity. Surgery , 106, 563-6. PMID: 2772830
- ↑ Tinckler LF. (1968). Triple appendix vermiformis--a unique case. Br J Surg , 55, 79-81. PMID: 5635427
- ↑ Canbay E & Akman E. (2011). Appendix perforation in appendix duplication in a man: a case report. J Med Case Rep , 5, 162. PMID: 21513538 DOI.
- ↑ 29.0 29.1 29.2 Levitt MA & Peña A. (2007). Anorectal malformations. Orphanet J Rare Dis , 2, 33. PMID: 17651510 DOI.
Reviews
Martin V & Shaw-Smith C. (2010). Review of genetic factors in intestinal malrotation. Pediatr. Surg. Int. , 26, 769-81. PMID: 20549505 DOI.
Strouse PJ. (2004). Disorders of intestinal rotation and fixation ("malrotation"). Pediatr Radiol , 34, 837-51. PMID: 15378215 DOI.
Levy AD & Hobbs CM. (2004). From the archives of the AFIP. Meckel diverticulum: radiologic features with pathologic Correlation. Radiographics , 24, 565-87. PMID: 15026601 DOI.
Chitkara DK, Nurko S, Shoffner JM, Buie T & Flores A. (2003). Abnormalities in gastrointestinal motility are associated with diseases of oxidative phosphorylation in children. Am. J. Gastroenterol. , 98, 871-7. PMID: 12738470 DOI.
D'Agostino J. (2002). Common abdominal emergencies in children. Emerg. Med. Clin. North Am. , 20, 139-53. PMID: 11826631
Boudiaf M, Soyer P, Terem C, Pelage JP, Maissiat E & Rymer R. (2001). Ct evaluation of small bowel obstruction. Radiographics , 21, 613-24. PMID: 11353110 DOI.
Articles
Mammadov E. (2018). Patent Omphalomesenteric Duct with Protruding Bowels through a Ruptured Omphalocele. Balkan Med J , 35, 118-119. PMID: 29400311 DOI.
Jones KL, Benirschke K & Chambers CD. (2009). Gastroschisis: etiology and developmental pathogenesis. Clin. Genet. , 75, 322-5. PMID: 19419414 DOI.
Feldkamp ML, Carey JC & Sadler TW. (2007). Development of gastroschisis: review of hypotheses, a novel hypothesis, and implications for research. Am. J. Med. Genet. A , 143A, 639-52. PMID: 17230493 DOI.
Cassart M, Massez A, Lingier P, Absil AS, Donner C & Avni F. (2006). Sonographic prenatal diagnosis of malpositioned stomach as a feature of uncomplicated intestinal malrotation. Pediatr Radiol , 36, 358-60. PMID: 16465538 DOI.
Ashraf A, Abdullatif H, Hardin W & Moates JM. (2005). Unusual case of neonatal diabetes mellitus due to congenital pancreas agenesis. Pediatr Diabetes , 6, 239-43. PMID: 16390394 DOI.
Beaudoin S, Mathiot-Gavarin A, Gouizi G & Bargy F. (2005). Familial malrotation: report of three affected siblings. Pediatr. Surg. Int. , 21, 856-7. PMID: 16205928 DOI.
Drewett M, Michailidis GD & Burge D. (2006). The perinatal management of gastroschisis. Early Hum. Dev. , 82, 305-12. PMID: 16563666 DOI.
Vegunta RK, Wallace LJ, Leonardi MR, Gross TL, Renfroe Y, Marshall JS, Cohen HS, Hocker JR, Macwan KS, Clark SE, Ramiro S & Pearl RH. (2005). Perinatal management of gastroschisis: analysis of a newly established clinical pathway. J. Pediatr. Surg. , 40, 528-34. PMID: 15793730 DOI.
Salomon LJ, Mahieu-Caputo D, Jouvet P, Jouannic JM, Benachi A, Grebille AG, Dumez Y & Dommergues M. (2004). Fetal home monitoring for the prenatal management of gastroschisis. Acta Obstet Gynecol Scand , 83, 1061-4. PMID: 15488122 DOI.
Langer JC. (2003). Abdominal wall defects. World J Surg , 27, 117-24. PMID: 12557047 DOI.
Search PubMed
Search Pubmed: gastrointestinal tract abnormalities | intestinal malrotation | Situs Inversus Viscera | Gastroschisis | Intestinal Aganglionosis
External Links
External Links Notice - The dynamic nature of the internet may mean that some of these listed links may no longer function. If the link no longer works search the web with the link text or name. Links to any external commercial sites are provided for information purposes only and should never be considered an endorsement. UNSW Embryology is provided as an educational resource with no clinical information or commercial affiliation.
- NIH National Institute of Diabetes and Digestive and Kidney Diseases Hirschsprung Disease | Anatomic Problems of the Colon
- BMC Gastroenterology
Glossary Links
- Glossary: A | B | C | D | E | F | G | H | I | J | K | L | M | N | O | P | Q | R | S | T | U | V | W | X | Y | Z | Numbers | Symbols | Term Link
Cite this page: Hill, M.A. (2024, June 18) Embryology Gastrointestinal Tract - Abnormalities. Retrieved from https://embryology.med.unsw.edu.au/embryology/index.php/Gastrointestinal_Tract_-_Abnormalities
- © Dr Mark Hill 2024, UNSW Embryology ISBN: 978 0 7334 2609 4 - UNSW CRICOS Provider Code No. 00098G