Renal System - Abnormalities
| Embryology - 26 Jul 2026 |
|---|
| Google Translate - select your language from the list shown below (this will open a new external page) |
|
العربية | català | 中文 | 中國傳統的 | français | Deutsche | עִברִית | हिंदी | bahasa Indonesia | italiano | 日本語 | 한국어 | မြန်မာ | Pilipino | Polskie | português | ਪੰਜਾਬੀ ਦੇ | Română | русский | Español | Swahili | Svensk | ไทย | Türkçe | اردو | ייִדיש | Tiếng Việt These external translations are automated and may not be accurate. (More? About Translations) |
| ICD-11 Structural developmental anomalies of the urinary system |
|---|
|
LB30 Structural developmental anomalies of kidneys - LB30.0 Renal agenesis or other reduction defects of kidney | LB30.1 Renal dysplasia | LB30.2 Congenital single renal cyst | LB30.3 Renal tubular dysgenesis | LB30.4 Oligomeganephronia | LB30.5 Accessory kidney | LB30.6 Fusion anomaly of kidneys | LB30.7 Ectopic or pelvic kidney | LB30.8 Medullary sponge kidney | LB30.9 Multicystic renal dysplasia LB31 Structural developmental anomalies of urinary tract - LB31.0 Congenital hydronephrosis | LB31.1 Congenital primary megaureter | LB31.2 Foetal lower urinary tract obstruction | LB31.3 Exstrophy of urinary bladder | LB31.4 Congenital diverticulum of urinary bladder | LB31.5 Duplication of urethra | LB31.6 Congenital megalourethra | LB31.7 Megacystis-megaureter | LB31.8 Atresia or stenosis of ureter | LB31.9 Agenesis of ureter | LB31.A Duplication of ureter | LB31.B Malposition of ureter | LB31.C Congenital absence of bladder or urethra | LB31.D Congenital vesico-uretero-renal reflux KC01 Congenital renal failure A severe irreversible decline in the ability of kidneys to remove wastes, concentrate urine, and maintain electrolyte balance; blood pressure; and calcium metabolism which existed at, or often before, birth. GB41 Nephrotic syndrome "A condition characterised by severe proteinuria, greater than 3.5 g/day in an average adult. The substantial loss of protein in the urine results in hypoalbuminaemia and generalised oedema. There is also usually hyperlipidaemia. Other manifestations of glomerular disease may be present. There are many possible causes and renal histological appearances. Possible complications include vascular thrombosis, infections, malnutrition and renal failure. |
Introduction

There are many different forms of renal development abnormalities associated with kidney, ureters, bladder and urethra. There are many genetic disorders associated with failure or abnormal renal development.
Prenatal diagnosis of obstructive and renal agenesis/dysgenesis disorders are also important for early reproductive decisions by the parents. For example, with bilateral renal agenesis, failure of both kidneys to development, is not compatible with fetal/neonatal survival.
"Horseshoe" kidney is the historic name for renal fusion, where the two kidneys are joined together usually by a single pole.
Because of their close developmental association, often described as the urogenital system, there can be an associated genital abnormalities.
There are also a range of branchiootorenal spectrum disorders (branchiootorenal syndrome and branchiootic syndrome) where both renal and auditory development are affected (for review see [1]).
Postnatally, newborn oxidative stress is thought to be involved in the progression of renal failure.
| System Abnormalities | ||||
|---|---|---|---|---|
|
Some Recent Findings

|
| More recent papers |
|---|
 This table allows an automated computer search of the external PubMed database using the listed "Search term" text link.
More? References | Discussion Page | Journal Searches | 2019 References | 2020 References Search term: Abnormal Renal Development | Renal Fusion | Horseshoe Kidney | Polycystic Kidney Disease | Congenital Nephrotic Syndrome] Triad Syndrome | |
| Older papers |
|---|
| These papers originally appeared in the Some Recent Findings table, but as that list grew in length have now been shuffled down to this collapsible table.
See also the Discussion Page for other references listed by year and References on this current page.
|
Australian Statistics

Statistics - Top Ten

The ten most frequently reported birth defects in Victoria between 2003-2004.
- Hypospadias
- Obstructive Defects of the Renal Pelvis or Obstructive Genitourinary Defects
- Ventricular Septal Defect
- Congenital Dislocated Hip
- Trisomy 21 or Down syndrome
- Hydrocephalus
- Cleft Palate
- Trisomy 18 or Edward Syndrome - multiple abnormalities of the heart, diaphragm, lungs, kidneys, ureters and palate 86% discontinued.
- Renal Agenesis/Dysgenesis - reduction in neonatal death and stillbirth since 1993 may be due to the more severe cases being identified in utero and being represented amongst the increased proportion of terminations (approximately 31%).
- Cleft Lip and Palate - occur with another defect in 33.7% of cases.
Obstructive Renal Pelvis Defect

Obstructive Renal Pelvis Defect (obstructive defects of the renal pelvis, uteropelvic junction obstruction, pelvo-uterero junction obstruction) is a term describing a developmental renal abnormality due to partial or complete blockage of the drainage of the kidney pelvis requiring surgical correction.
The blockage during development can be due to failure of recanalization of the outflow tract.
The blockage can have several anatomical causes including:
The blockage leads to an accumulation of urine in the affected region, with several potential effects: nephron damage from compression (hydronephrosis); decreased urine output leading to lack of amniotic fluid (oligohydramnios); respiratory development effects due to the lack of amniotic fluid. The most common type of obstruction is at the uteropelvic junction (UPJ), between the junction of the ureter and the kidney. Blockage lower as the ureter enters the bladder, the ureterovesicular junction (UVJ), usually involves only one kidney and the back flow enlarges the affected ureter (megaureter).
Renal Agenesis or Dysgenesis
| International Classification of Diseases - Renal Agenesis or Dysgenesis Anomalies | ||
|---|---|---|
|
Potter's syndrome or sequence is a rare condition (1 in 2000-5000) resulting in bilateral renal agenesis and occurs more frequently in primigravid mothers and more commonly in male offspring. At birth affected infants have a characteristic facial phenotype known as "Potter facies", named after Potter (1946) original description.[8]

Unilateral renal absence is relatively common and may be asymptomatic. In the complete form, bilateral absence, the child is not viable and dies within a few days of birth.
Features associated with this anomaly are:
- Oligohydramnios
- Amnion nodosum (small warty amnion with accretions of squamous cells on the inner wall). This is tangible evidence for oligohydramnios.
- Facial deformities: This results from uterine moulding around the head. The ears are low slung and simple, the mandible is small, the nose flattened and the eyes exhibit Pre-epicanthic folds. This is a horseshoe shaped flap of skin from the upper lid to the cheek in front of the epicanthus. (Downs syndrome has an epicanthic fold). Note that the genesis is occasionally incomplete allowing survival (e.g.) Causal factors are largely unknown although there is some familial predisposition. There has been described a mutation in the enzyme, heparan sulfate 2-sulfotransferase, that generates a similar phenotype in the mouse.
Note that upper G.I.T. obstruction is associated with POLYHYDRAMNIOS whereas failure of fetal micturition is associated with OLIGOHYDRAMNIOS with consequent firm uterine moulding on the fetus, leading to facial, locomotor and palatal deformities.

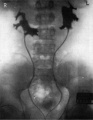
Renal agenesis. I.V. pyelography showing right renal agenesis (or dysplasia) (>). The right hip dysplasia is also shown (>>).[10]
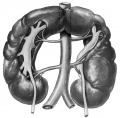
Renal Fusion

| ICD-11 LB30.62 Horseshoe kidney - Horseshoe kidney is the most frequent renal fusion anomaly and is characterised by the union of the inferior poles of the two kidneys through an isthmus. Horseshoe kidney is in fact an anatomical anomaly rather than a disease, but it does lead to predisposition to certain conditions such as hydronephrosis, nephrolithiasis or pyelonephritis. One third of individuals with horseshoe kidney are asymptomatic, with the anomaly being discovered fortuitously during a radiological examination. Urogenital or renal vessel anomalies may be associated with the condition. For cases requiring treatment, various therapeutic approaches are available and choice of treatment depends on the associated pathology. |
The less common renal fusions are LB30.60 Lobulated kidney and LB30.61 Fused pelvic kidney.
Also described as Horseshoe kidney.
|

Renal fusion (pathology specimen) |
Historic
Eisendrath DN Phifer FM and Culver HB. Horseshoe Kidney (1925) Ann Surg. 82(5): 735-64. PubMed 17865363

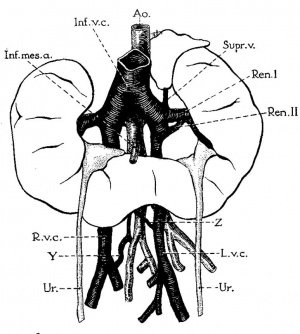
Horseshoe kidney with symmetric halves
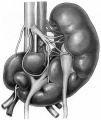
Horseshoe kidneys with asymmetric halves.
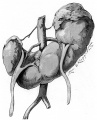
Horseshoe kidneys with asymmetric halves.
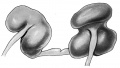
Well marked fibrous isthmus joining the two halves.
Horseshoe kidney with superior isthmus.
Note unusual form of both pelves.






Boyden EA. Description of a horseshoe kidney associated with left inferior vena cava and disc-shaped suprarenal glands, together with a note on the occurrence of horseshoe kidneys in human embryos. (1931) Anat. Rec. 51(2): 187-211.

Fig. 1 Horseshoe kidney
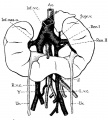
Fig. 2 Horseshoe kidney
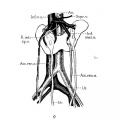
Fig. 9 Horseshoe kidney
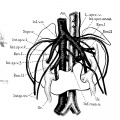
Fig. 10 Horseshoe kidney




Renal-Adrenal Fusion
A rare anomaly of the upper pole of the kidney historically first described by Rokitansky in 1855[11]. Intrarenal ectopic adrenal tissue can also occur and in such instances consist of adrenal cortical tissue with no adrenal medullary tissue. Other organ fusions can occur with splenogonadal fusion the most commonly reported.
Triad Syndrome

Also described as Prune Belly Syndrome.
The Triad is:
- Agenesis of abdominal wall muscles
- Bladder outflow obstruction
- Bilateral undescended testes
The condition was first described by Frolich[12]and then called "prune belly syndrome" as a descriptive, because the intestinal pattern is evident through the thin protruding abdominal wall in the infant.[13]
Survival of the prune belly child depends on the number of functioning remaining nephrons at birth and the operability of the obstruction.
In some cases there are vestiges of muscle in the abdominal wall and it is not known whether this represents (a) destruction of muscle, or (b) failure of development of muscle. The causes of this malformation are little known, but maternal therapy with estrogens in the first trimester has been implicated frequently.

Hydronephrosis

Renal outflow obstruction
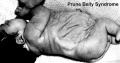
Prune belly
Polycystic Kidney Disease


- diffuse cystic malformation of both kidneys
- cystic malformations of liver and lung often associated, Often familial disposition
- Two types
- Infantile (inconsistent with prolonged survival)
- Adult (less severe and allows survival)
- Autosomal dominant PKD disease - recently identified at mutations in 2 different human genes encoding membrane proteins (possibly channels)
|

Cyst formation at the level of the cell, nephron, and kidney[15] |
Nephroblastoma (Wilms' Tumor)

- (nephroblastoma) Named after Max Wilms, a German doctor who wrote first medical articles 1899
- most common type of kidney cancer children
- WT1 gene - encodes a zinc finger protein[16][17]
- Both constitutional and somatic mutations disrupting the DNA-binding domain of WT1 result in a potentially dominant-negative phenotype
- some blastema cells (mass of undifferentiated cells) persist to form a ‘nephrogenic rest’
- Most rests become dormant or regress but others proliferate to form hyperplastic rests
- any type of rest can then undergo a genetic or epigenetic change to become a neoplastic rest
- can proliferate further to produce a benign lesion (adenomatous rest) or a malignant Wilms’ tumour
Renal Cysts
The Bosniak classification system (I - IV) was designed to separate identified cystic renal masses by analysis of computed tomography (CT) features into surgical and nonsurgical categories.[18] Named after Morton Bosniak, Yale University School of Medicine, the developer of this classification system.
- Category I lesions are simple benign cysts showing homogeneity, water content, and a sharp interface with adjacent renal parenchyma, with no wall thickening, calcification, or enhancement.
- Category II consists of cystic lesions with one or two thin (<=1 mm thick) septations or thin, fine calcification in their walls or septa (wall thickening > 1 mm advances the lesion into surgical category III) and hyperdense benign cysts with all the features of category I cysts except for homogeneously high attenuation. A benign category II lesion must be 3 cm or less in diameter, have one quarter of its wall extending outside the kidney so the wall can be assessed, and be nonenhancing after contrast material is administered.
- Category IIF consists of minimally complicated cysts that need followup. This is a group not well defined by Bosniak but consists of lesions that do not neatly fall into category II. These lesions have some suspicious features that deserve followup to detect any change in character.
- Category III consists of true indeterminate cystic masses that need surgical evaluation, although many prove to be benign. They may show uniform wall thickening, nodularity, thick or irregular peripheral calcification, or a multilocular nature with multiple enhancing septa. Hyperdense lesions that do not fulfill category II criteria are included in this group.
- Category IV lesions with a nonuniform or enhancing thick wall, enhancing or large nodules in the wall, or clearly solid components in the cystic lesion. Enhancement was considered present when lesion components increased by at least 10 H.
Urorectal Septum Malformation
Urorectal septum malformation thought to be a deficiency in caudal mesoderm which in turn leads to the malformation of the urorectal septum and other structures in the pelvic region. Recent research has also identified the potential presence of a persistent urachus prior to septation of the cloaca (common urogenital sinus).
Clinically the condition is described as a urorectal septum malformation sequence (URSMS): female disorder of sexual development; ambiguous external genitalia; imperforate anus, vagina, and urethra; renal, colonic, and lumbosacral anomalies.
Renal Vascular Anomalies
There is an excellent recent 2010 review[19] of renal vascular anomalies shown in adults using computed tomography. The images below are from that review.
Renal Arteries

|

|
| Multiple renal arteries | Accessory renal artery |
Renal Veins

Supernumerary right renal vein |

Supernumerary right renal vein |

Multiple right renal veins |

Multiple right renal veins |
Renal Vascular Anomalies: Multiple renal arteries | Accessory renal artery | Supernumerary right renal vein 1 | Supernumerary right renal vein 1 | Multiple right renal veins 2 | Multiple right renal veins 2 | Cardiovascular System Development
Bladder Exstrophy
| ICD-11 LB31.3 Exstrophy of urinary bladder - Bladder exstrophy (or classic bladder exstrophy) is a congenital genitourinary malformation belonging to the spectrum of the exstrophy-epispadias complex and is characterized by an evaginated bladder plate, epispadias and an anterior defect of the pelvis, pelvic floor and abdominal wall. |

- developmental abnormality associated with bladder development.
- origins appear to occur not just by abnormal bladder development, but by a congenital malformation of the ventral wall of abdomen (between umbilicus and pubic symphysis).
- There may also be other anomolies associated with failure of closure of abdominal wall and bladder (epispadias, pubic bone anomolies).
Bladder
- absent or small bladder - associated with renal agenesis.
Bladder Duplication

Bladder duplication |
An extremely rare abnormality. This Magnetic Resonance Image (MRI) shows a complete bladder duplication with urethra duplication, diphallus, anorectal malformation and rightsided renal agensis with ipsilateral gonadal agenesisis.[20]
|
Ureterocele
The distal ureter balloons at the opening into the bladder and forms a sac-like pouch. Often associated with other ureter abnormalities.
Ureter and Urethra
| International Classification of Diseases - Ureter and Urethra Anomalies | ||
|---|---|---|
|

- Ureter - Duplex Ureter, ectopic ureter
- Meyer-Weigert Rule - clinical term for the arrangement of the ureter in a completely duplicated renal system, the ureter from the upper renal pole enters the bladder more medially and caudally than the ureter from the lower renal pole.
- Urethra - Urethral Obstruction and Hypospadias

Ectopic and dilated left ureter (small arrows) inserting ectopically into the obstructed left hemivagina (asterisk) on MR imaging in a 17-year-old girl.[21]
Male lower urinary tract obstruction can occur with posterior urethral valves (PUV) (incidence 1.6 to 2.1 per 10 000 births), an anterior urethral valves (AUV) are 25–30‐fold less common.[22]
| ICD-11 LB31.2 Foetal lower urinary tract obstruction - A disease caused by partial or complete obstruction of the urethra, during the antenatal period. This disease can present with enlarged bladder, oligohydramnios, or pulmonary hypoplasia. Confirmation is through observation of the obstruction by imaging. |
See also Genital System - Abnormalities
Nephrotic Syndrome

(Congenital Nephrotic syndrome, lipoid nephrosis)
| GB41 Nephrotic syndrome A condition characterised by severe proteinuria, greater than 3.5 g/day in an average adult. The substantial loss of protein in the urine results in hypoalbuminaemia and generalised oedema. There is also usually hyperlipidaemia. Other manifestations of glomerular disease may be present. There are many possible causes and renal histological appearances. Possible complications include vascular thrombosis, infections, malnutrition and renal failure. |
Genetic Mutations include:
- nephrin - protein of the slit diaphragm of renal filtration barrier, located at the cell surface in the area between two podocytes. NPHS1 gene location 19q13.12, mutations in this gene are associated with Congenital Nephrotic Syndrome (Nephrotic syndrome). (More? renal abnormalities)
- podocin - protein of the slit diaphragm of renal filtration barrier, located at the cell surface in the area between two podocytes. NPHS2 gene location 1q25.2, mutations in this gene are associated with Congenital Nephrotic Syndrome (Nephrotic syndrome). (More? renal abnormalities)
References
- ↑ Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A & Smith RJH. (1993). Branchiootorenal Spectrum Disorders. , , . PMID: 20301554
- ↑ 2.0 2.1 Fogelgren B, Polgar N, Lui VH, Lee AJ, Tamashiro KK, Napoli JA, Walton CB, Zuo X & Lipschutz JH. (2015). Urothelial Defects from Targeted Inactivation of Exocyst Sec10 in Mice Cause Ureteropelvic Junction Obstructions. PLoS ONE , 10, e0129346. PMID: 26046524 DOI.
- ↑ Avanoglu A & Tiryaki S. (2020). Embryology and Morphological (Mal)Development of UPJ. Front Pediatr , 8, 137. PMID: 32318525 DOI.
- ↑ Mikuz G. (2018). Ectopias of the kidney, urinary tract organs, and male genitalia. Pathologe , , . PMID: 30446779 DOI.
- ↑ Nef S, Neuhaus TJ, Spartà G, Weitz M, Buder K, Wisser J, Gobet R, Willi U & Laube GF. (2016). Outcome after prenatal diagnosis of congenital anomalies of the kidney and urinary tract. Eur. J. Pediatr. , 175, 667-76. PMID: 26805407 DOI.
- ↑ Halachmi S & Pillar G. (2008). Congenital urological anomalies diagnosed in adulthood - management considerations. J Pediatr Urol , 4, 2-7. PMID: 18631884 DOI.
- ↑ Lu BC, Cebrian C, Chi X, Kuure S, Kuo R, Bates CM, Arber S, Hassell J, MacNeil L, Hoshi M, Jain S, Asai N, Takahashi M, Schmidt-Ott KM, Barasch J, D'Agati V & Costantini F. (2009). Etv4 and Etv5 are required downstream of GDNF and Ret for kidney branching morphogenesis. Nat. Genet. , 41, 1295-302. PMID: 19898483 DOI.
- ↑ POTTER EL. (1946). Facial characteristics of infants with bilateral renal agenesis. Am. J. Obstet. Gynecol. , 51, 885-8. PMID: 20984673
- ↑ 9.0 9.1 Alorainy IA, Barlas NB & Al-Boukai AA. (2010). Pictorial Essay: Infants of diabetic mothers. Indian J Radiol Imaging , 20, 174-81. PMID: 21042439 DOI.
- ↑ Acién P, Galán F, Manchón I, Ruiz E, Acién M & Alcaraz LA. (2010). Hereditary renal adysplasia, pulmonary hypoplasia and Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome: a case report. Orphanet J Rare Dis , 5, 6. PMID: 20388228 DOI.
- ↑ Rokitansky K. A manual of pathologic anatomy. Vol II. Philadelphia, Pa: Blanchard & Lea, 1855; 188.
- ↑ Frolich, F. Der Mangel der Muskeln, insbesondere der Seitenbauchmuskeln. Dissertation: Wurzburg (pub.) 1839.
- ↑ Osler, W. Congenital absence of the abdominal muscles with distended and hypertrophied urinary bladder. Bull. Johns Hopkins Hosp. 12: 331-333, 1901.
- ↑ Kumar M, Gupta U, Thakur S, Aggrawal S, Meena J, Sharma S & Trivedi SS. (2012). Prenatal sonographic evaluation and postnatal outcome of renal anomalies. Indian J Hum Genet , 18, 75-82. PMID: 22754226 DOI.
- ↑ Chapin HC & Caplan MJ. (2010). The cell biology of polycystic kidney disease. J. Cell Biol. , 191, 701-10. PMID: 21079243 DOI.
- ↑ Call KM, Glaser T, Ito CY, Buckler AJ, Pelletier J, Haber DA, Rose EA, Kral A, Yeger H & Lewis WH. (1990). Isolation and characterization of a zinc finger polypeptide gene at the human chromosome 11 Wilms' tumor locus. Cell , 60, 509-20. PMID: 2154335
- ↑ Sharma PM, Yang X, Bowman M, Roberts V & Sukumar S. (1992). Molecular cloning of rat Wilms' tumor complementary DNA and a study of messenger RNA expression in the urogenital system and the brain. Cancer Res. , 52, 6407-12. PMID: 1330293
- ↑ Israel GM & Bosniak MA. (2005). How I do it: evaluating renal masses. Radiology , 236, 441-50. PMID: 16040900 DOI.
- ↑ Kumar S, Neyaz Z & Gupta A. (2010). The utility of 64 channel multidetector CT angiography for evaluating the renal vascular anatomy and possible variations: a pictorial essay. Korean J Radiol , 11, 346-54. PMID: 20461189 DOI.
- ↑ Gajbhiye V, Nath S, Ghosh P, Chatterjee A, Haldar D & Das SK. (2015). Complete duplication of the urinary bladder: An extremely rare congenital anomaly. Urol Ann , 7, 91-3. PMID: 25657554 DOI.
- ↑ Wang ZJ, Daldrup-Link H, Coakley FV & Yeh BM. (2010). Ectopic ureter associated with uterine didelphys and obstructed hemivagina: preoperative diagnosis by MRI. Pediatr Radiol , 40, 358-60. PMID: 19924410 DOI.
- ↑ Murakami T, Asanuma H, Shigeta K, Ezaki T & Oya M. (2018). Rare association of the anterior and posterior urethral valves. Pediatr Int , 60, 1090-1091. PMID: 30585406 DOI.
- ↑ Scott RP & Quaggin SE. (2015). Review series: The cell biology of renal filtration. J. Cell Biol. , 209, 199-210. PMID: 25918223 DOI.
Reviews
Chariatte V, Ramseyer P & Cachat F. (2013). Uroradiological screening for upper and lower urinary tract anomalies in patients with hypospadias: a systematic literature review. Evid Based Med , 18, 11-20. PMID: 22815315 DOI.
Woodhouse CR, Neild GH, Yu RN & Bauer S. (2012). Adult care of children from pediatric urology. J. Urol. , 187, 1164-71. PMID: 22335866 DOI.
Articles
Sinha R, Vasudevan A, Agarwal I, Sethi SK, Saha A, Pradhan S, Ekambaram S, Thaker N, Matnani M, Banerjee S, Sharma J, Singhal J, Ashraf S & Mandal K. (2020). Congenital Nephrotic Syndrome in India in the Current Era: A Multicenter Case Series. Nephron , 144, 21-29. PMID: 31655822 DOI.
Fernando MA, Creighton SM & Wood D. (2015). The long-term management and outcomes of cloacal anomalies. Pediatr. Nephrol. , 30, 759-65. PMID: 25217327 DOI.
Chowdhary SK, Kandpal DK, Sibal A & Srivastava RN. (2014). Management of complicated ureteroceles: Different modalities of treatment and long-term outcome. J Indian Assoc Pediatr Surg , 19, 156-61. PMID: 25197194 DOI.
Search Pubmed
Search Pubmed: Obstructive Renal Pelvis Defects | Renal Agenesis | hydronephrosis | Ureterocele
Historic
Vermooten V. Congenital cystic dilatation of the renal collecting tubules: A new disease entity. (1951) Yale J Biol Med. 23(6): 450–453. PMID 14836770
Smith E. and Strasberg A. Cysts of the genital ducts, Müllerian and Wolffian. (1946) Can Med Assoc J. 55(2): 119–121. PMID 20323857
Rusche CF and Bacon SK. Congenital renal anomalies: with special reference to horseshoe kidney. (1939) Cal West Med. 50: 344-348. PMID 18745137
Randall A. The origin and growth of renal calculi. (1937) Ann Surg. 105(6): 1009–1027. PMID 18745137
Morton JJ. and Jones TB. Obstructions about the mesentery in infants. (1936) Ann. Surg. 104: 864-891. PMID 17856875
Bell ET. Cystic disease of the kidneys. (1935) Am J Pathol. 11(3): 373–418. PMID 1910969
Oppenheimer GD. Polycystic disease of the kidney. (1934) Ann Surg. 100(6): 1136–1158. PMID 17856426
Abbott 1930 Anomalies of the genito-urinary tract. (1930) J. Can. Med. Assoc. -226.
Papin E. and Eisendrath DN. Classification of renal and ureteral anomalies. (1927) Ann Surg. 85(5): 735–756. PMID 17865673
Piersol GM. Polycystic disease of the kidney. (1927) Trans Am Climatol Clin Assoc , 43, 221-231. PMID 21408945
Hinman F. Gibson TE. and Kutzmann AA. Cysts of the Wolffian body. (1924) Ann Surg. 79(5): 762–769. PMID 17865035
Eisendrath DN. Congenital strictures and spiral twists of the ureters. (1917) Ann Surg. 65(5): 552–559. PMID17863705
Braasch WF. VIII. The clinical diagnosis of congenital anomaly in the kidney and ureter. (1912) 56, 726-737. PMID17862922
Eisendrath DN. Congenital malformations of the ureters. (1912) Ann Surg. 55(4): 571–592. PMID17862830
Spicer JE. Dystocia due to distension of the urinary bladder of the fœtus, with remarks on renal secretion in utero. (1909) Proc R Soc Med. 2(Obstet Gynaecol Sect): 1–24. PMID 19973800
Hamann CA. Spindle-shaped dilatations and tortuosity of the ureters in the fetus. (1902) J Med Res. 8(1): 125–127. PMID19971486
Terms
- anterior urethral valve - uncommon real abnormality.
- bilateral renal agenesis - (BRA) failure of both kidney development and is not compatible with life and is associated with oligohydramnios and pulmonary hypoplasia.
- CAKUT - congenital anomalies of the kidneys and urinary tract.
- Congenital Nephrotic Syndrome - (CNS, Nephrotic syndrome) rare kidney disorder characterized by heavy proteinuria, hypoproteinemia, and edema starting soon after birth. Most cases are caused by genetic abnormalities in the components of the glomerular filtration barrier, especially nephrin and podocin.
- cystic dysplasia - term used in renal dysplasia if cysts are present.
- Meyer-Weigert Rule - clinical term for the arrangement of the ureter in a completely duplicated renal system, the ureter from the upper renal pole enters the bladder more medially and caudally than the ureter from the lower renal pole.
- nephrin - protein of the slit diaphragm of renal filtration barrier, located at the cell surface in the area between two podocytes. NPHS1 gene location 19q13.12.
- Nephrotic Syndrome - (Congenital Nephrotic syndrome, CNS) rare kidney disorder characterized by heavy proteinuria, hypoproteinemia, and edema starting soon after birth. Most cases are caused by genetic abnormalities in the components of the glomerular filtration barrier, especially nephrin and podocin.
- podocin - protein of the slit diaphragm of renal filtration barrier, located at the cell surface in the area between two podocytes. NPHS2 gene location 1q25.2.
- posterior urethral valve - (congenital obstructing posterior urethral membrane) a common male renal abnormality (~1 in 10,000-25,000 live births) classified into 3 types. Membrane from mesonephric (Wolffian) duct origin forms of a thick, valve-like structure.
- renal agenesis - term for a complete absence of renal development.
- renal dysgenesis - term for several forms of abnormal kidney development, which includes aplasia, hypoplasia, dysplasia, and cystic disease.
- renal aplasia - term for a rudimentary kidney without any functional nephrons.
- renal hypoplasia - term for a small non-dysplastic kidney that has less than the normal number of calyces and nephrons.
- renal dysplasia - term for focal, diffuse, or segmentally arranged primitive structures, specifically primitive ducts, resulting from abnormal metanephric differentiation. There can also be non-renal elements present.
- renal adysplasia - term is used when aplasia and severe dysplasia are aspects of phenotypic spectrum.
- unilateral renal agenesis - (URA) failure of a single kidney development, often asymptomatic if not detected by prenatal screening.
| Renal Terms | ||
|---|---|---|
| ||
|
{kind=link}
{kind=link}
{kind=link}
Glossary Links
- Glossary: A | B | C | D | E | F | G | H | I | J | K | L | M | N | O | P | Q | R | S | T | U | V | W | X | Y | Z | Numbers | Symbols | Term Link
Cite this page: Hill, M.A. (2026, July 26) Embryology Renal System - Abnormalities. Retrieved from https://embryology.med.unsw.edu.au/embryology/index.php/Renal_System_-_Abnormalities
- © Dr Mark Hill 2026, UNSW Embryology ISBN: 978 0 7334 2609 4 - UNSW CRICOS Provider Code No. 00098G