BGDB Sexual Differentiation - Abnormalities
Reproductive Abnormalities

|
This page introduces the statistical data associated with birth abnormalities in the genital system in Australia and also briefly introduces some of these defects. For more detailed coverage look at the Genital Abnormalities Notes. Abnormalites associated with development of secondary sex characteristics including mammary development are not covered in this current practical class.
|
Disorders of Sex Development
The previous human sex development terminology (true hermaphrodites, male pseudohermaphrodites and female pseudohermaphrodites) are considered outdated and stigmatising and have been replaced with the general term Disorders of Sex Development (DSD) established by the Consensus statement on management of intersex disorders.[1] See also the Medical Journal of Australia 2009 editorial article.[2]
See DSD Terminology
Sex Chromosomes
| Trisomy X | Klinefelter syndrome | Monosomy X Turner Syndrome |
|---|---|---|

|

|

|
| Trisomy X is a caused by the presence of an extra X chromosome in females (47,XXX instead of 46,XX). This is also the most common female chromosomal abnormality, occurring in approximately 1 in 1,000 female births. Most females with triple X syndrome have normal sexual development. | Klinefelter syndrome (47,XXY or XXY) in males has several features including sterility. See also 2011 Science student project. | Turner Syndrome Monosomy X is a chromosomal disorder (approximately 1 in 2000 live female births) caused by a complete or partial X monosomy in some or all cells. Features sexual development altered during puberty, early end to menstrual cycles not due to pregnancy, and inability to conceive without fertility treatment. See also 2011 Science student project. |
Critical Periods
This figure provides a broad general summary of key events in genital development in relation to critical periods.

Gonadal Descent

|
Cryptorchidism
|

|
Hernia
Hydrocele
|
External Genitalia

|
Penoscrotal Hypospadias 3D Ultrasound
Ultrasonography in rendering mode, at GA 33 weeks, with short penis and with evidence of testicles inside a bifid scrotum. |

|
Hypospadias
Opening of the urethra (meatus) on the inferior surface (ring) not at the tip. Other regions where urethra may open to the surface (dashed line). |
- Links: hypospadias
Internal Genitalia
Male
- Ductus Deferens - unilateral or bilateral absence congenital unilateral absence of the vas deferens (CUAVD), Congenital bilateral absence of the vas deferens (CBAVD)
- failure of mesonephric duct to differentiate frequent cause of obstructive azoospermia
- 75% of men with bilateral absence have at least one detectable common mutation in the cystic fibrosis transmembrane conductance regulator (CFTR) gene
Female
- Uterus - Uterine Duplication, Unicornate Uterus, Septate Uterus.
- Cervical - cervical agenesis, cervical duplication
- Vagina Absence - Failure of sinovaginal bulb development, 1 in 4,000 to 5,000 female births.
- MRI Female Genital and Ureter Abnormality
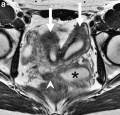
Uterine didelphys
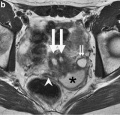
Two cervices
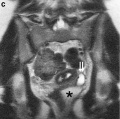
Obstructed hemivagina



| Ultrasound Bicornuate Uterus | ||||
|---|---|---|---|---|
|
This ultrasound scan shows a bicornuate uterus and an ectopic (cornual) pregnancy. |

This cartoon shows the new 2013 ESHRE-ESGE classification system[3] based on uterine anatomy, embryological origin is the secondary basic characteristic, and both cervical and vaginal anomalies are classified in independent co-existent sub-classes. For more detail see Additional Information.
Other Abnormalities
Use the links to find out more about these topics.
- congenital adrenal hyperplasia - impairment of cortisol production by the adrenal cortex, is one of the most common causes of intersex genitalia at birth.
- Androgen Insensitivity Syndrome - genetically male (XY) unable to respond to male sex hormones (androgens), they may have mostly female sex characteristics or signs of both male and female sexual development. Also a partial androgen insensitivity syndrome (PAIS) associated with impaired male genital development that can be transmitted through mutations in the androgen receptor.
- Cryptorchidism - covered in detail above.
- Undescended Ovaries - reasonably rare gonad abnormality, often detected following clinical assessment of fertility problems and may also be associated with other uterine malformations (unicornuate uterus).
- Hydrocele - covered above, most commonly male (but can also be female) fluid-filled cavity.
DSD Terminology
| New Terminology | Previous Terminology |
|---|---|
| DSD | Intersex |
| 46,XY DSD | Male pseudohermaphrodite undervirilization of an XY male undermasculinization of an XY male |
| 46,XX DSD | Female pseudohermaphrodite overvirilization of an XX female masculinization of an XX female |
| Ovotesticular DSD | True hermaphrodite |
| 46,XX testicular DSD | XX male or XX sex reversal |
| 46,XY complete gonadal dysgenesis | XY sex reversal |
Abnormalities Interactive Component
| Attempt the Quiz - Abnormalities | ||
|---|---|---|

Here are a few simple Quiz questions that relate to Abnormalities of genital development from the lecture and practical.
|
Additional Information
| Additional Information - Content shown under this heading is not part of the material covered in this class. It is provided for those students who would like to know about some concepts or current research in topics related to the current class page. |
DSD Differential Diagnosis
| Contribution of AMH to DSD differential diagnosis[4] | |
|---|---|

|
|
Trisomy X
| ICD-11 Karyotype 47,XXX - Trisomy X is a sex chromosome anomaly with a variable phenotype caused by the presence of an extra X chromosome in females (47,XXX instead of 46,XX). Most individuals are only mildly affected or asymptomatic, the most common physical features including tall stature, epicanthal folds, hypotonia and clinodactyly, with seizures, renal and genitourinary abnormalities, and premature ovarian failure being also associated findings. |
This is also the most common female chromosomal abnormality, occurring in approximately 1 in 1,000 female births with some individuals are only mildly affected or asymptomatic. It is estimated that only 10% of individuals with trisomy X are actually diagnosed. Common physical features include: tall stature, epicanthal folds, hypotonia and clinodactyly. Other physical features include: neural (seizures), renal and genitourinary abnormalities, and premature ovarian failure (POF) (see review[5].
Turner Syndrome
| ICD-11 Turner syndrome - Karyotype missing one X chromosome (45, X0 or 45,XO/46,XX mosaicism) ; gonads: ovaries (streak); phenotype female with short stature, amenorrhea (hypergonadotrophic hypogonadism), absence of sexual development, webbed neck, low set ears, posterior hairline, widely-spaced nipples, short fourth metacarpals, and increased carrying angle at the elbow (cubitus valgus). Often associated with renal, cardiac and ocular abnormalities. |
Turner syndrome or Monosomy X (45,X) feature include gonadal dysgenesis and short stature. Named after Henry Turner, an American clinician who first described (1938) the syndrome.

Turner syndrome karyotype (45,X)
Klinefelter Syndrome
| ICD-11 Klinefelter syndrome - Klinefelter syndrome defines a group of chromosomal disorders in which there is at least one extra X chromosome compared with the normal 46,XY male karyotype. The effects on physical features and on physical and cognitive development increase with the number of extra X's, and each extra X is associated with an intelligence quotient (IQ) decrease of approximately 15-16 points, with language most affected, particularly expressive language skills. |
Klinefelter syndrome (47,XXY) caused by an additional X chromosome (or more) in affected males> Named after Harry F. Klinefelter who first described (1942) the syndrome. Common physical features include reduced fertility and hypogonadism. Some individuals are only mildly affected or asymptomatic and the severity varies greatly between individuals.
International Classification of Diseases
Hypospadias
| Hypospadia Classification | Meatus Opening |
| Anterior | on inferior surface of glans penis |
| Coronal | in balanopenile furrow |
| Distal | on distal third of shaft |
| Penoscrotal | at base of shaft in front of scrotum |
| Scrotal | on scrotum or between the genital swellings |
| Perineal | behind scrotum or genital swellings |
| Links: Genital Abnormalities | Penis Development | |
- epispadias - Uncommon abnormality associated with the penis, 1 in 30,000 infant males, external urethral opening on the dorsal surface of penis.
- cryptorchidism in common eutherian mammals.[6] - Species comparison of descent timeline
- Mayer-Rokitansky syndrome (MRK anomaly, Rokitansky-Küster-Hauser syndrome, RKH syndrome, RKH) congenital absence of the vagina, dyspareunia, vaginal agenesis.
{kind=link}
Uterine Anomalies
| ESHRE/ESGE Classification of Uterine Anomalies | |
|---|---|
European Society of Human Reproduction and Embryology (ESHRE) and the European Society for Gynaecological Endoscopy (ESGE)
|
|
| <pubmed>23894234</pubmed>
See also ICD10 Congenital malformations of genital organs (Q50-Q56) | |
{kind=link}
- Uterine Duplication (uterus didelphys, double uterus, uterus didelphis) A rare uterine developmental abnormality where the paramesonephric ducts (Mullerian ducts) completely fail to fuse generating two separate uterus parts each connected to the cervix and having an ovary each. Failure of fusion of lower paramesonephric ducts, with either double or single vagina.
- Unicornate Uterus - failure of the paramesonephric ducts to fuse. A single paramesomnephric duct has fused with the vaginal plate and now opens into the vagina, while the other forms a diverticulum.
- Septate Uterus
Polycystic Ovary Syndrome
(PCOS) or Stein–Leventhal syndrome (1930s researchers) a metabolic syndrome with many other symptoms, ovarian cysts arise through incomplete follicular development or failure of ovulation. For review see[7]
Congenital Adrenal Hyperplasia
| Congenital Adrenal Hyperplasia | |||
|---|---|---|---|
| Type | Enzyme Deficiency | Female | Male |
| classic virilizing adrenal hyperplasia | 21-hydroxylase, 11-beta-hydroxylase, or 3-beta-hydroxysteroid dehydrogenase |
ambiguous genitalia at birth - complete or partial fusion of the labioscrotal folds and a phallic urethra to clitoral enlargement (clitoromegaly), partial fusion of the labioscrotal folds, or both | normal genitalia, present at age 1-4 weeks with salt wasting (salt-wasting adrenal hyperplasia) |
| simple virilizing adrenal hyperplasia | mild 21-hydroxylase | identified later in childhood because of precocious pubic hair, clitoral enlargement (clitoromegaly), or both, often accompanied by accelerated growth and skeletal maturation | early genital development (pubic hair and/or phallic enlargement) accelerated growth and skeletal maturation |
| nonclassic adrenal hyperplasia | milder deficiencies of 21-hydroxylase or 3-beta-hydroxysteroid dehydrogenase |
present at puberty or adult with infrequent menstruation (oligomenorrhea), abnormal hair growth (hirsutism), and/or infertility | |
| 17-hydroxylase deficiency syndrome | 17-hydroxylase deficiency or 3-beta-hydroxysteroid dehydrogenase |
rare, phenotypically female at birth do not develop breasts or menstruate in adolescence and may have hypertension | steroidogenic acute regulatory (StAR) deficiency have ambiguous genitalia or female genitalia, at puberty may lack breast development and may have hypertension |
| Prader Stages | |
|---|---|
| Stage 0 | Normal female genitalia. |
| Stage 1 | Mildly enlarged clitoris, slightly reduced vaginal opening, usually within normal variations. |
| Stage 2 | Abnormal genitalia clearly seen by eye, phallus being intermediate in size, small vaginal opening with separate urethral opening. Posterior labial fusion present. |
| Stage 3 | Further enlarged phallus than Stage 2, with single urogenital sinus and nearly complete fusion of the labia. |
| Stage 4 | Upon examination, looks more male than female, with an empty scrotum and a normal-sized penis-like phallus, however this structure is not quite as free from the perineum to be pulled onto the abdomen towards the umbilicus. A small urethral/vaginal opening at the base of the shaft/phallus (hypospadias in a male), with an x-ray showing the internal connection with the upper vagina and uterus. |
| Stage 5 | Complete male virilisation, a normally-formed penis is present. Urethral opening at or near the tip, and the scrotum formed, but empty. Internal organs in the pelvis include normal ovaries and uterus, with the vagina connecting internally with the urethra (as in Stage 4). Newborn infants are not seen to be visibly ambiguous, and are assumed to be normal boys (with undescended testes). The diagnosis of CAH is not apparent until signs of salt-wasting develop, about a week later. |
| Stage 6 | Normal male presentation of the penis with normal testes. |
| Links: Genital - CAH | Adrenal - CAH | Female | Genital System - Abnormalities | Genital Terms | |
Persistent Mullerian Duct Syndrome
| Rare mutations in the AMH gene or its receptor can lead to persistence of the entire, or parts of, the paramesonephric duct (Mullerian Duct) as shown in this surgical image.[8] | 
|
Abnormalities Sex Ratios
| Male preponderance | Female preponderance |
|---|---|
|
|
| Cardiac defects | |
|
|
| Table data[9] Links: abnormal development | cardiovascular abnormalities | USA | Male | Female | cleft lip and palate | |
Environmental Abnormalities
Template:Diethylstilbestrol (DES or diethylstilbetrol) - is a drug that was prescribed to women from 1938-1971 to prevent miscarriage in high-risk pregnancies.
- The drug acted as a potent estrogen (mimics natural hormone) and therefore could also act as a potential endocrine disruptor.
- This led to a number of developing fetal reproductive tract and other abnormalities.
- In the female fetus, it increased risk of abnormal reproductive tract and also carcinogenic (cancer forming).
- In the male fetus, it increased the occurance of abnormal genitalia.
- The drug was banned by FDA (USA) in 1979 as a teratogen, it had previously also been used as livestock growth promoter and could have potentially entered the human food chain.
Sex Development Genetics
The table below indicates a number of genes that are involved in sexual development. Mutations in these genes can also lead to a number of different known genital abnormalities.
| Gene (OMIM) | Protein Function | Gonad Phenotype of Null Mice | Human Syndrome | |
| Bipotential gonad | ||||
| Wt1 | Transcription factor | Blockage in genital ridge development | Denys-Drash, WAGR, Frasier syndrome | |
| Sf1 | Nuclear receptor | Blockage in genital ridge development | Embryonic testicular regression syndrome | |
| Lhx9 | Transcription factor | Blockage in genital ridge development | a | |
| Emx2 | Transcription factor | Blockage in genital ridge development | a | |
| M33 | Transcription factor | Gonadal dysgenesis | a | |
| Testis-determining pathway | ||||
| Gata4/Fog2 | Transcription/cofactor | Reduced Sry levels, XY sex reversal | a | |
| Sry | Transcription factor | XY sex reversal | XY sex reversal (LOF); XX sex reversal (GOF) | |
| Sox9 | Transcription factor | XY sex reversal | Campomelic dysplasia, XX sex reversal (GOF) | |
| Sox8 | Transcription factor | XY sex reversal in combination with partial loss of Sox9 function | a | |
| Fgf9 | Signaling molecule | XY sex reversal | a | |
| Dax1 | Nuclear receptor | Impaired testis cord formation and spermatogenesis | Hypogonadism | |
| Pod1 | Transcription factor | XY sex reversal | a | |
| Dhh | Signaling molecule | Impaired differentiation of Leydig and PM cells | XY gonadal dysgenesis | |
| Pgdra | Receptor | Reduction in mesonephric cell migration | a | |
| Pgds | Enzyme | No phenotype | a | |
| Arx | Transcription factor | Abnormal testicular differentiation | X-linked lissencephaly with abnormal genitalia | |
| Atrx | Helicase | ND | ATRX syndrome | |
| Insl3 | Signaling factor | Blockage of testicular descent | Cryptorchidism | |
| Lgr8 | Receptor | Blockage of testicular descent | Cryptorchidism | |
| Hoxa10 | Transcription factor | Blockage of testicular descent | Cryptorchidism | |
| Hoxa11 | Transcription factor | Blockage of testicular descent | Cryptorchidism | |
| Amh | Hormone | No Müllerian duct degeneration | Persistent Müllerian duct syndrome | |
| Misrl1 | Receptor | No Müllerian duct degeneration | Persistent Müllerian duct syndrome | |
| Pax2 | Transcription factor | Dysgenesis of mesonephric tubules | a | |
| Lim1 | Transcription factor | Agenesis of Wolffian and Müllerian ducts | a | |
| Dmrt1 | Transcription factor | Loss of Sertoli and germ cells | XY femaleb | |
| Ovary-determining pathway | ||||
| Wnt4 | Signaling molecule | Müllerian duct agenesis, testosterone synthesis, and coelomic vessel formation | XY female (GOF) | |
| FoxL2 | Transcription factor | Premature ovarian failure | BPES | |
| Dax1 | Nuclear receptor | XY sex reversal (GOF) | XY sex reversal (GOF) | |
| RSPO1 | Signaling molecule | XX sex reversal (LOF) | XX sex reversal (LOF) | |
| Table Legend | ||||
|
a No mutations in human sexual disorders identified to date.
b Candidate gene for 9p deletion, XY sex reversal. | |||
| Table data modified[10] | ||||
References
- ↑ Lee PA, Houk CP, Ahmed SF & Hughes IA. (2006). Consensus statement on management of intersex disorders. International Consensus Conference on Intersex. Pediatrics , 118, e488-500. PMID: 16882788 DOI.
- ↑ Warne GL & Hewitt JK. (2009). Disorders of sex development: current understanding and continuing controversy. Med. J. Aust. , 190, 612-3. PMID: 19485836
- ↑ Grimbizis GF, Gordts S, Di Spiezio Sardo A, Brucker S, De Angelis C, Gergolet M, Li TC, Tanos V, Brölmann H, Gianaroli L & Campo R. (2013). The ESHRE-ESGE consensus on the classification of female genital tract congenital anomalies. Gynecol Surg , 10, 199-212. PMID: 23894234 DOI.
- ↑ Xu HY, Zhang HX, Xiao Z, Qiao J & Li R. (2019). Regulation of anti-Müllerian hormone (AMH) in males and the associations of serum AMH with the disorders of male fertility. Asian J. Androl. , 21, 109-114. PMID: 30381580 DOI.
- ↑ Tartaglia NR, Howell S, Sutherland A, Wilson R & Wilson L. (2010). A review of trisomy X (47,XXX). Orphanet J Rare Dis , 5, 8. PMID: 20459843 DOI.
- ↑ Amann RP & Veeramachaneni DN. (2007). Cryptorchidism in common eutherian mammals. Reproduction , 133, 541-61. PMID: 17379650 DOI.
- ↑ Norman RJ, Wu R & Stankiewicz MT. (2004). 4: Polycystic ovary syndrome. Med. J. Aust. , 180, 132-7. PMID: 14748678
- ↑ Ren X, Wu D & Gong C. (2017). Persistent Müllerian duct syndrome: A case report and review. Exp Ther Med , 14, 5779-5784. PMID: 29285121 DOI.
- ↑ Michalski AM, Richardson SD, Browne ML, Carmichael SL, Canfield MA, VanZutphen AR, Anderka MT, Marshall EG & Druschel CM. (2015). Sex ratios among infants with birth defects, National Birth Defects Prevention Study, 1997-2009. Am. J. Med. Genet. A , 167A, 1071-81. PMID: 25711982 DOI.
- ↑ Wilhelm D, Palmer S & Koopman P. (2007). Sex determination and gonadal development in mammals. Physiol. Rev. , 87, 1-28. PMID: 17237341 DOI.
| Genital System Terms (expand to view) |
|---|
Note there are additional glossaries associated with spermatozoa, oocyte renal.
|
| Other Terms Lists |
|---|
| Terms Lists: ART | Birth | Bone | Cardiovascular | Cell Division | Endocrine | Gastrointestinal | Genital | Genetic | Head | Hearing | Heart | Immune | Integumentary | Neonatal | Neural | Oocyte | Palate | Placenta | Radiation | Renal | Respiratory | Spermatozoa | Statistics | Tooth | Ultrasound | Vision | Historic | Drugs | Glossary |
BGDB: Lecture - Gastrointestinal System | Practical - Gastrointestinal System | Lecture - Face and Ear | Practical - Face and Ear | Lecture - Endocrine | Lecture - Sexual Differentiation | Practical - Sexual Differentiation | Tutorial
Glossary Links
- Glossary: A | B | C | D | E | F | G | H | I | J | K | L | M | N | O | P | Q | R | S | T | U | V | W | X | Y | Z | Numbers | Symbols | Term Link
Cite this page: Hill, M.A. (2026, April 20) Embryology BGDB Sexual Differentiation - Abnormalities. Retrieved from https://embryology.med.unsw.edu.au/embryology/index.php/BGDB_Sexual_Differentiation_-_Abnormalities
- © Dr Mark Hill 2026, UNSW Embryology ISBN: 978 0 7334 2609 4 - UNSW CRICOS Provider Code No. 00098G