Talk:Cardiovascular System - Abnormalities
| About Discussion Pages |
|---|

Glossary Links
Cite this page: Hill, M.A. (2026, August 1) Embryology Cardiovascular System - Abnormalities. Retrieved from https://embryology.med.unsw.edu.au/embryology/index.php/Talk:Cardiovascular_System_-_Abnormalities |
2019
First trimester combined screening biochemistry in detection of congenital heart defects
J Matern Fetal Neonatal Med. 2019 Oct;32(19):3272-3277. doi: 10.1080/14767058.2018.1462324. Epub 2018 Apr 22.
Alanen J1, Korpimaki T2, Kouru H2, Sairanen M2, Leskinen M3, Gissler M4, Ryynanen M1, Nevalainen J1.
Objective: To evaluate the performance of first trimester biochemical markers, pregnancy-associated plasma protein-A (PAPP-A), free beta human chorionic gonadotropin (fβ-hCG), and nuchal translucency (NT) in detection of severe congenital heart defects (CHDs). Methods: During the study period from 1 January 2008 to 31 December 2011, biochemical markers and NT were measured in 31,144 women as part of voluntary first trimester screening program for Down's syndrome in Northern Finland. Data for 71 severe CHD cases and 762 controls were obtained from the hospital records and from the National Medical Birth Register, which records the birth of all liveborn and stillborn infants, and from the National Register of Congenital Malformations that receives information about all the CHD cases diagnosed in Finland. Results: Both PAPP-A and fβ-hCG multiple of median (MoM) values were decreased in all severe CHDs: 0.71 and 0.69 in ventricular septal defects (VSDs), 0.58 and 0.88 in tetralogy of Fallot cases (TOFs), 0.82 and 0.89 in hypoplastic left heart syndromes (HLHSs), and 0.88 and 0.96 in multiple defects, respectively. NT was increased in all study groups except of VSD group. ROC AUC was 0.72 for VSD when combining prior risk with PAPP-A and fβ-hCG. Adding NT did not improve the detection rate. With normal NT but decreased (<0.5 MoM) PAPP-A and fβ-hCG odds ratios for VSD and HLHS were 19.5 and 25.6, respectively. Conclusions: Maternal serum biochemistry improves the detection of CHDs compared to NT measurement only. In cases with normal NT measurement but low concentrations of both PAPP-A and fβ-hCG, an alert for possible CHD, especially VSD, could be given with thorough examination of fetal heart in later ultrasound scans. KEYWORDS: Congenital heart defect; first trimester screening; nuchal translucency PMID: 29683008 DOI: 10.1080/14767058.2018.1462324
Association of NKX2-5, GATA4, and TBX5 polymorphisms with congenital heart disease in Egyptian children
Mol Genet Genomic Med. 2019 Mar 4:e612. doi: 10.1002/mgg3.612. [Epub ahead of print]
Behiry EG1, Al-Azzouny MA1, Sabry D2, Behairy OG3, Salem NE4.
BACKGROUND: Several genes encoding transcription factors are known to be the primary cause of congenital heart disease. NKX2-5 and GATA4 were the first congenital heart disease-causing genes identified by linkage analysis. This study designed to study the association of five single-nucleotide variants of NKX2-5, GATA4, and TBX5 genes with sporadic nonsyndromic cases of a congenital cardiac septal defect in Egyptian children. METHODS: Venous blood samples from 150 congenital heart disease children (including a ventricular septal defect, atrial septal defect, tetralogy of Fallot, and patent ductus arteriosus) and 90 apparently healthy of matched age and sex were studied by polymerase chain reaction followed by direct sequencing in order to study two single-nucleotide variants of NKX2-5 (rs2277923, rs28936670), two single-nucleotide variants of GATA4 (rs368418329, rs56166237) and one single-nucleotide variant TBX5 (rs6489957). The distribution of genotype and allele frequency in the congenital heart diseases (CHD) group and control group were analyzed. RESULTS: We found different genotype frequencies of the two variants of NKX2-5, as CT genotype of rs2277923 was present in 58% and 36% in cases and control respectively, and TT genotype present in 6% of the cases. Also regarding missense variant rs28936670, heterozygous AG presented in 82% of the cases. Also, we observed a five prime UTR variant rs368418329, GT (42% of the cases) and GG (46% of the cases) genotypes showed the most frequent presentation in cases. While regarding a synonymous variant rs56166237, GT and GG were the most presented in cases (41.4%, 56% respectively) in contrast to control group (20%, 1.7% respectively). Also, a synonymous variant in TBX5, the distribution of genotype frequency was significantly different between the CHD group and control group. CT genotype of TBX5 -rs6489957 was found in 12 ASD, 24 VSD, six PDA, three aortic coarctation and nine fallot that represent 42% of the cases. CONCLUSIONS: Significantly higher frequency of different allelle of five variants was observed in cases when compared to the control group, with significant risky effect for the development of septal defect. In addition to two polymorphisms of NKX2-5 (rs2277923, rs28936670) variant in the cardiac septal defect, two variants in GATA4 (rs368418329, rs56166237) and one variant in TBX5 (rs6489957) seem to have a role in the pathogenesis of congenital heart disease. © 2019 The Authors. Molecular Genetics & Genomic Medicine published by Wiley Periodicals, Inc. KEYWORDS: GATA4 ; NKX2-5 ; TBX5 ; Congenital heart disease PMID: 30834692 DOI: 10.1002/mgg3.612
2018
Early fetal ultrasound screening for major congenital heart defects without Doppler
Eur J Obstet Gynecol Reprod Biol. 2018 Dec 14;233:93-97. doi: 10.1016/j.ejogrb.2018.11.030.
García Fernández S1, Arenas Ramirez J2, Otero Chouza MT2, Rodriguez-Vijande Alonso B2, Llaneza Coto ÁP3.
Abstract
OBJECTIVE: Congenital heart defects are the most common major structural fetal abnormalities. Color flow mapping has played a dominant role in the detection of abnormalities during the first trimester, regardless of the International Society of Ultrasound in Obstetrics and Gynecology warning on the use of Doppler during early pregnancy. The aim of our study was to investigate the use of transvaginal two-dimensional sonography without Doppler for assessing the four-chamber view and the outflow tract view of fetuses at 11-13 weeks of gestation for cardiac screening of major congenital heart defects.
STUDY DESIGN: This was a prospective observational study conducted in the Fetal Medicine Unit of Cabueñes University Hospital, between May 2014 and August 2015. Only low risk-pregnancies were studied. All ultrasonographic examinations were performed by two experienced sonographers in maternal-fetal medicine. The combination of high-frequency transvaginal (nine MHz) and transabdominal (six MHz) ultrasonography transducers were used. An early cardiac screening was performed in 97% of cases. Statistical analysis was carried out using successive multivariate logistic regression models in order to investigate the effect of crown-rump length and body mass index on the probability of visualizing the four-chamber view and/or the outflow tract view.
RESULTS: 663 low-risk pregnant women were included. Regarding the transvaginal approach, neither the crown-rump length nor the body mass index had a statistically significant relationship on the probability of visualization of the four-chamber view and outflow tract view. For the transabdominal approach, the crown-rump length and the body mass index presented a statistically significant effect on the visualization of the four-chamber view and the outflow tract view. Using the transvaginal approach: the success rate of performing a four-chamber view was 89.4% and 82.4% for the outflow tract view. Using the transabdominal approach: the success rate of performing a four-chamber view was 77.8% and 61.5% for the outflow tract view. Four major congenital heart defects were diagnosed, and the prenatal ultrasonagraphic diagnosis was confirmed for all cases.
CONCLUSIONS: Routine first-trimester ultrasonagraphy without Doppler, when performed by experienced sonographers, can effectively identify major congenital heart defects. Additional multicenter well designed studies should clarify the feasibility of this approach.
Copyright © 2018. Published by Elsevier B.V.
KEYWORDS: Congenital heart defects; Doppler ultrasound; Early fetal ultrasonography; First trimester; Safe ultrasound PMID: 30580230 DOI: 10.1016/j.ejogrb.2018.11.030
Utility of fetal cardiac magnetic resonance imaging to assess fetuses with right aortic arch and right ductus arteriosus
J Matern Fetal Neonatal Med. 2018 Jun;31(12):1627-1631. doi: 10.1080/14767058.2017.1322951. Epub 2017 May 7.
Dong SZ1, Zhu M1.
Abstract
OBJECTIVE: To evaluate the utility of fetal cardiac magnetic resonance imaging (MRI) to diagnose right aortic arch (RAA) with right ductus arteriosus. METHODS: This retrospective study included six fetuses with right aortic arch and right ductus arteriosus. The six fetal cases were examined using a 1.5-T magnetic resonance unit. The steady-state free precession (SSFP) and single-shot turbo spin echo (SSTSE) sequences were used to evaluate the fetal heart and airway. The gestational age of the six fetuses ranged from 22 to 35 weeks (mean, 26.5 weeks). The age of the pregnant women ranged from 23 to 40 years (mean 31 years). RESULTS: Fetal cardiac MRI diagnosed the six fetal cases with RAA with right ductus arteriosus correctly. Among the six fetuses, four were associated with other congenital heart defects. In three of six cases, the diagnoses established using prenatal echocardiography (echo) was correct when compared with postnatal diagnosis. CONCLUSIONS: Fetal cardiac MRI is a useful complementary tool to assess fetuses with RAA and right ductus arteriosus. KEYWORDS: Fetus; cardiac magnetic resonance; right aortic arch; right ductus arteriosus
PMID: 28438064 DOI: 10.1080/14767058.2017.1322951
2017
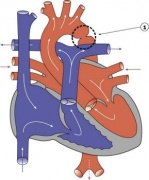
International Pediatric Cardiac Code
International Paediatric and Congenital Cardiac Code
Report from the international society for nomenclature of paediatric and congenital heart disease: creation of a visual encyclopedia illustrating the terms and definitions of the international pediatric and congenital cardiac code
World J Pediatr Congenit Heart Surg. 2010 Oct;1(3):300-13. doi: 10.1177/2150135110379622.
Giroud JM1, Jacobs JP, Spicer D, Backer C, Martin GR, Franklin RC, Béland MJ, Krogmann ON, Aiello VD, Colan SD, Everett AD, William Gaynor J, Kurosawa H, Maruszewski B, Stellin G, Tchervenkov CI, Walters HL 3rd, Weinberg P, Anderson RH, Elliott MJ.
Abstract
Tremendous progress has been made in the field of pediatric heart disease over the past 30 years. Although survival after heart surgery in children has improved dramatically, complications still occur, and optimization of outcomes for all patients remains a challenge. To improve outcomes, collaborative efforts are required and ultimately depend on the possibility of using a common language when discussing pediatric and congenital heart disease. Such a universal language has been developed and named the International Pediatric and Congenital Cardiac Code (IPCCC). To make the IPCCC more universally understood, efforts are under way to link the IPCCC to pictures and videos. The Archiving Working Group is an organization composed of leaders within the international pediatric cardiac medical community and part of the International Society for Nomenclature of Paediatric and Congenital Heart Disease (www.ipccc.net). Its purpose is to illustrate, with representative images of all types and formats, the pertinent aspects of cardiac diseases that affect neonates, infants, children, and adults with congenital heart disease, using the codes and definitions associated with the IPCCC as the organizational backbone. The Archiving Working Group certifies and links images and videos to the appropriate term and definition in the IPCCC. These images and videos are then displayed in an electronic format on the Internet. The purpose of this publication is to report the recent progress made by the Archiving Working Group in establishing an Internet-based, image encyclopedia that is based on the standards of the IPCCC. KEYWORDS: International Pediatric and Congenital Cardiac Code; Internet; cardiac encyclopedia; cardiac images; cardiac nomenclature; congenital heart disease; databases PMID 23804886 DOI: 10.1177/2150135110379622
Nomenclature for congenital and paediatric cardiac disease: historical perspectives and The International Pediatric and Congenital Cardiac Code
Cardiol Young. 2008 Dec;18 Suppl 2:70-80. doi: 10.1017/S1047951108002795.
Franklin RC1, Jacobs JP, Krogmann ON, Béland MJ, Aiello VD, Colan SD, Elliott MJ, William Gaynor J, Kurosawa H, Maruszewski B, Stellin G, Tchervenkov CI, Walters Iii HL, Weinberg P, Anderson RH.
Abstract
Clinicians working in the field of congenital and paediatric cardiology have long felt the need for a common diagnostic and therapeutic nomenclature and coding system with which to classify patients of all ages with congenital and acquired cardiac disease. A cohesive and comprehensive system of nomenclature, suitable for setting a global standard for multicentric analysis of outcomes and stratification of risk, has only recently emerged, namely, The International Paediatric and Congenital Cardiac Code. This review, will give an historical perspective on the development of systems of nomenclature in general, and specifically with respect to the diagnosis and treatment of patients with paediatric and congenital cardiac disease. Finally, current and future efforts to merge such systems into the paperless environment of the electronic health or patient record on a global scale are briefly explored. On October 6, 2000, The International Nomenclature Committee for Pediatric and Congenital Heart Disease was established. In January, 2005, the International Nomenclature Committee was constituted in Canada as The International Society for Nomenclature of Paediatric and Congenital Heart Disease. This International Society now has three working groups. The Nomenclature Working Group developed The International Paediatric and Congenital Cardiac Code and will continue to maintain, expand, update, and preserve this International Code. It will also provide ready access to the International Code for the global paediatric and congenital cardiology and cardiac surgery communities, related disciplines, the healthcare industry, and governmental agencies, both electronically and in published form. The Definitions Working Group will write definitions for the terms in the International Paediatric and Congenital Cardiac Code, building on the previously published definitions from the Nomenclature Working Group. The Archiving Working Group, also known as The Congenital Heart Archiving Research Team, will link images and videos to the International Paediatric and Congenital Cardiac Code. The images and videos will be acquired from cardiac morphologic specimens and imaging modalities such as echocardiography, angiography, computerized axial tomography and magnetic resonance imaging, as well as intraoperative images and videos. Efforts are ongoing to expand the usage of The International Paediatric and Congenital Cardiac Code to other areas of global healthcare. Collaborative efforts are underway involving the leadership of The International Nomenclature Committee for Pediatric and Congenital Heart Disease and the representatives of the steering group responsible for the creation of the 11th revision of the International Classification of Diseases, administered by the World Health Organisation. Similar collaborative efforts are underway involving the leadership of The International Nomenclature Committee for Pediatric and Congenital Heart Disease and the International Health Terminology Standards Development Organisation, who are the owners of the Systematized Nomenclature of Medicine or "SNOMED". The International Paediatric and Congenital Cardiac Code was created by specialists in the field to name and classify paediatric and congenital cardiac disease and its treatment. It is a comprehensive code that can be freely downloaded from the internet (http://www.IPCCC.net) and is already in use worldwide, particularly for international comparisons of outcomes. The goal of this effort is to create strategies for stratification of risk and to improve healthcare for the individual patient. The collaboration with the World Heath Organization, the International Health Terminology Standards Development Organisation, and the healthcare industry, will lead to further enhancement of the International Code, and to its more universal use. PMID 19063777 DOI: 10.1017/S1047951108002795 [PubMed - indexed for MEDLINE]
2016
Long-Term Survival of Individuals Born With Congenital Heart Disease: A Systematic Review and Meta-Analysis
J Am Heart Assoc. 2016 Jun 16;5(6). pii: e002846. doi: 10.1161/JAHA.115.002846.
Best KE1, Rankin J2.
Abstract
BACKGROUND: Estimates of long-term survival are required to adequately assess the variety of health and social services required by those with congenital heart disease (CHD) throughout their lives. METHODS AND RESULTS: Medline, Embase, and Scopus were searched from inception to June 2015 using MeSH headings and keywords. Population-based studies that ascertained all persons born with CHD within a predefined area and reported survival estimates at ≥5 years were included. Unadjusted survival estimates for each CHD subtype at ages 1 year, 5 years, 10 years, and so forth were extracted. Pooled survival estimates for each age were calculated using meta-analyses. Metaregression was performed to examine the impact of study period on survival. Of 7840 identified articles, 16 met the inclusion criteria. Among those with CHD, pooled 1-year survival was 87.0% (95% CI 82.1-91.2), pooled 5-year survival was 85.4% (95% CI 79.4-90.5), and pooled 10-year survival was 81.4% (95% CI 73.8-87.9). There was significant heterogeneity of survival estimates among articles (P<0.001 for 1-, 5-, and 10-year survival). A more recent study period was significantly associated with greater survival at ages 1 year (P=0.047), 5 years (P=0.013), and 10 years (P=0.046). Survival varied by CHD subtype, with 5-year survival being greatest for those with ventricular septal defect (96.3%, 95% CI 93.7-98.2) and lowest for those with hypoplastic left heart (12.5%, 95% CI 0.0-41.4). CONCLUSIONS: Among persons with CHD, the mortality rate is greatest during the first year of life; however, this systematic review and meta-analysis showed that survival decreases gradually after infancy and into adulthood. © 2016 The Authors. Published on behalf of the American Heart Association, Inc., by Wiley Blackwell. KEYWORDS: congenital; heart defects; survival
PMID 27312802
De Novo and Rare Variants at Multiple Loci Support the Oligogenic Origins of Atrioventricular Septal Heart Defects
PLoS Genet. 2016 Apr 8;12(4):e1005963. doi: 10.1371/journal.pgen.1005963. eCollection 2016.
Priest JR1,2, Osoegawa K3, Mohammed N4, Nanda V5, Kundu R6, Schultz K4, Lammer EJ4, Girirajan S7, Scheetz T8, Waggott D6, Haddad F2,6, Reddy S1,2, Bernstein D1,2, Burns T9, Steimle JD10, Yang XH10, Moskowitz IP10, Hurles M11, Lifton RP12,13, Nickerson D14, Bamshad M14,15, Eichler EE13,14, Mital S16, Sheffield V13,17, Quertermous T2,6, Gelb BD18, Portman M15, Ashley EA2,6. Author information Abstract Congenital heart disease (CHD) has a complex genetic etiology, and recent studies suggest that high penetrance de novo mutations may account for only a small fraction of disease. In a multi-institutional cohort surveyed by exome sequencing, combining analysis of 987 individuals (discovery cohort of 59 affected trios and 59 control trios, and a replication cohort of 100 affected singletons and 533 unaffected singletons) we observe variation at novel and known loci related to a specific cardiac malformation the atrioventricular septal defect (AVSD). In a primary analysis, by combining developmental coexpression networks with inheritance modeling, we identify a de novo mutation in the DNA binding domain of NR1D2 (p.R175W). We show that p.R175W changes the transcriptional activity of Nr1d2 using an in vitro transactivation model in HUVEC cells. Finally, we demonstrate previously unrecognized cardiovascular malformations in the Nr1d2tm1-Dgen knockout mouse. In secondary analyses we map genetic variation to protein-interaction networks suggesting a role for two collagen genes in AVSD, which we corroborate by burden testing in a second replication cohort of 100 AVSDs and 533 controls (p = 8.37e-08). Finally, we apply a rare-disease inheritance model to identify variation in genes previously associated with CHD (ZFPM2, NSD1, NOTCH1, VCAN, and MYH6), cardiac malformations in mouse models (ADAM17, CHRD, IFT140, PTPRJ, RYR1 and ATE1), and hypomorphic alleles of genes causing syndromic CHD (EHMT1, SRCAP, BBS2, NOTCH2, and KMT2D) in 14 of 59 trios, greatly exceeding variation in control trios without CHD (p = 9.60e-06). In total, 32% of trios carried at least one putatively disease-associated variant across 19 loci,suggesting that inherited and de novo variation across a heterogeneous group of loci may contribute to disease risk.
PMID 27058611
http://journals.plos.org/plosgenetics/article?id=10.1371/journal.pgen.1005963
2011
Congenital heart disease in the newborn requiring early intervention
Korean J Pediatr. 2011 May;54(5):183-91. Epub 2011 May 31.
Yun SW. Source Department of Pediatrics, Chung-Ang University College of Medicine, Seoul, Korea.
Abstract
Although antenatal diagnostic technique has considerably improved, precise detection and proper management of the neonate with congenital heart disease (CHD) is always a great concern to pediatricians. Congenital cardiac malformations vary from benign to serious conditions such as complete transposition of the great arteries (TGA), critical pulmonary and aortic valvular stenosis/atresia, hypoplastic left heart syndrome (HLHS), obstructed total anomalous pulmonary venous return (TAPVR), which the baby needs immediate diagnosis and management for survival. Unfortunately, these life threatening heart diseases may not have obvious evidence early after birth, most of the clinical and physical findings are nonspecific and vague, which makes the diagnosis difficult. High index of suspicion and astute acumen are essential to decision making. When patent ductus arteriosus (PDA) is opened widely, many serious malformations may not be noticed easily in the early life, but would progress as severe acidosis/shock/cyanosis or even death as PDA constricts after few hours to days. Ductus dependent congenital cardiac lesions can be divided into the ductus dependent systemic or pulmonary disease, but physiologically quite different from each other and treatment strategy has to be tailored to the clinical status and cardiac malformations. Inevitably early presentation is often regarded as a medical emergency. Differential diagnosis with inborn error metabolic disorders, neonatal sepsis, persistent pulmonary hypertension of the newborn (PPHN) and other pulmonary conditions are necessary. Urgent identification of the newborn at such high risk requires timely referral to a pediatric cardiologist, and timely intervention is the key in reducing mortality and morbidity. This following review deals with the clinical presentations, investigative modalities and approach to management of congenital cardiac malformations presenting in the early life.
PMID 21829408
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3145901
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Spontaneous Closure of Muscular Trabecular Ventricular Septal Defect: Comparison of Defect Positions
Acta Paediatr. 2011 Apr 22. doi: 10.1111/j.1651-2227.2011.02333.x. [Epub ahead of print] Miyake T, Shinohara T, Inoue T, Marutani S, Takemura T. Source Department of Pediatrics, Kinki University School of Medicine, Osakasayama, Japan. Abstract Aim: To evaluate the timing and frequency of spontaneous closure of the muscular trabecular ventricular septal defect (VSD). Methods: We performed a historical cohort study for which 150 patients <3 months of age (median age, 9 days) diagnosed as having a muscular trabecular VSD were selected. Median age at latest follow-up was 2.8 years. Another 32 patients diagnosed after 3 months of age were also reviewed. Using color Doppler, defects were classified into 3 groups: anterior, apical, and midventricular. Results: Spontaneous closure occurred in 126 patients (84%): anterior, 36 of 47 (83%); apical, 26 of 31 (84%); and midventricular, 64 of 72 (89%). Multivariate analyses showed a lower frequency of spontaneous closure for patients of age of ≥20 days at initial echocardiography (hazard ratio 0.60, 95% confidence interval [CI] 0.39 to 0.89) and for anterior and apical muscular trabecular VSD (hazard ratio 0.66, 95% CI 0.47 to 0.95). The prevalence of the midventricular muscular trabecular VSD was significantly lower in patients ≥3 months of age at initial echocardiography than in those <3 months (p = 0.010). Conclusion: We infer that midventricular muscular trabecular VSD tends to close spontaneously earlier and more frequently than either anterior or apical muscular trabecular VSD.
Acta Paediatrica © 2011 Foundation Acta Paediatrica.
PMID 21517965
Racial/Ethnic Disparities in Risk of Early Childhood Mortality Among Children With Congenital Heart Defects
Pediatrics. 2011 Apr 18. [Epub ahead of print]
Nembhard WN, Salemi JL, Ethen MK, Fixler DE, Dimaggio A, Canfield MA. Source Departments of Epidemiology and Biostatistics and. Abstract
Background: Infants with congenital heart defects (CHDs) have increased risk of childhood morbidity and mortality. However, little is known about racial/ethnic differences in early childhood mortality. Patients and Methods: We conducted a retrospective cohort study with data from the Texas Birth Defect Registry on 19 530 singleton, live-born infants with a CHD and born January 1, 1996, to December 31, 2003, to non-Hispanic (NH) white, NH black, and Hispanic women. Texas Birth Defect Registry data were linked to Texas death records and the National Death Index to ascertain deaths between January 1, 1996, and December 31, 2005. Kaplan-Meier survival estimates were computed, and hazard ratios (HRs) and 95% confidence intervals (CIs) were calculated from multivariable Cox-proportional hazard regression models to determine the effect of maternal race/ethnicity on mortality for selected CHD phenotypes. Results: After adjusting for covariates, compared with NH white children, NH black children had increased early childhood mortality risk for transposition of the great arteries (HR: 2.04 [95% CI: 1.40-2.97]), tetralogy of Fallot (HR: 1.85 [95% CI: 1.09-3.12]), pulmonary valve atresia without ventricular septal defect (VSD) (HR: 2.60 [95% CI: 1.32-5.12]), VSD (HR: 1.56 [95% CI: 1.19-2.03]), and atrial septal defect (HR: 1.34 [95% CI: 1.08-1.66]). Hispanic children had higher mortality risk for pulmonary valve atresia without VSD (HR: 1.76 [95% CI: 1.06-2.91]) and hypoplastic left heart syndrome (HR: 1.51 [95% CI: 1.13-2.02]). Conclusions: We provide evidence that supports racial/ethnic disparities in early childhood mortality among infants with CHDs. Identifying infants with the greatest risk of early childhood mortality will facilitate development of interventions and policies to mitigate these risks.
PMID 21502234
2010
Beta-thalassemia
Orphanet J Rare Dis. 2010 May 21;5:11. doi: 10.1186/1750-1172-5-11.
Galanello R, Origa R. Source Dipartimento di Scienze Biomediche e Biotecnologie- Università di Cagliari, Ospedale Regionale, Microcitemie ASL Cagliari, Cagliari, Italy. renzo.galanello@mcweb.unica.it
Abstract
Beta-thalassemias are a group of hereditary blood disorders characterized by anomalies in the synthesis of the beta chains of hemoglobin resulting in variable phenotypes ranging from severe anemia to clinically asymptomatic individuals. The total annual incidence of symptomatic individuals is estimated at 1 in 100,000 throughout the world and 1 in 10,000 people in the European Union. Three main forms have been described: thalassemia major, thalassemia intermedia and thalassemia minor. Individuals with thalassemia major usually present within the first two years of life with severe anemia, requiring regular red blood cell (RBC) transfusions. Findings in untreated or poorly transfused individuals with thalassemia major, as seen in some developing countries, are growth retardation, pallor, jaundice, poor musculature, hepatosplenomegaly, leg ulcers, development of masses from extramedullary hematopoiesis, and skeletal changes that result from expansion of the bone marrow. Regular transfusion therapy leads to iron overload-related complications including endocrine complication (growth retardation, failure of sexual maturation, diabetes mellitus, and insufficiency of the parathyroid, thyroid, pituitary, and less commonly, adrenal glands), dilated myocardiopathy, liver fibrosis and cirrhosis). Patients with thalassemia intermedia present later in life with moderate anemia and do not require regular transfusions. Main clinical features in these patients are hypertrophy of erythroid marrow with medullary and extramedullary hematopoiesis and its complications (osteoporosis, masses of erythropoietic tissue that primarily affect the spleen, liver, lymph nodes, chest and spine, and bone deformities and typical facial changes), gallstones, painful leg ulcers and increased predisposition to thrombosis. Thalassemia minor is clinically asymptomatic but some subjects may have moderate anemia. Beta-thalassemias are caused by point mutations or, more rarely, deletions in the beta globin gene on chromosome 11, leading to reduced (beta+) or absent (beta0) synthesis of the beta chains of hemoglobin (Hb). Transmission is autosomal recessive; however, dominant mutations have also been reported. Diagnosis of thalassemia is based on hematologic and molecular genetic testing. Differential diagnosis is usually straightforward but may include genetic sideroblastic anemias, congenital dyserythropoietic anemias, and other conditions with high levels of HbF (such as juvenile myelomonocytic leukemia and aplastic anemia). Genetic counseling is recommended and prenatal diagnosis may be offered. Treatment of thalassemia major includes regular RBC transfusions, iron chelation and management of secondary complications of iron overload. In some circumstances, spleen removal may be required. Bone marrow transplantation remains the only definitive cure currently available. Individuals with thalassemia intermedia may require splenectomy, folic acid supplementation, treatment of extramedullary erythropoietic masses and leg ulcers, prevention and therapy of thromboembolic events. Prognosis for individuals with beta-thalassemia has improved substantially in the last 20 years following recent medical advances in transfusion, iron chelation and bone marrow transplantation therapy. However, cardiac disease remains the main cause of death in patients with iron overload.
PMID 20492708
Features and outcomes in utero and after birth of fetuses with myocardial disease
Int J Pediatr. 2010;2010:628451. Epub 2010 Oct 3.
Fesslova V, Mongiovì M, Pipitone S, Brankovic J, Villa L. Source Center of Fetal Cardiology, Policlinico San Donato IRCCS, Via Morandi 30, San Donato Milanese, 20097 Milan, Italy.
Abstract
Objectives. Ninety-one fetuses with dilated or hypertrophic cardiomyopathy (DCM, HCM) and myocarditis were studied. Results. Group 1 "DCM" included 19 fetuses: 13 with hydrops (FH) and 5 with associated extracardiac anomalies (ECAs) (15.8%). Group 2 "Myocarditis" included twelve fetuses, having 11 with FH. Group 3 "HCM" included sixty fetuses: 26 had associated ECAs, 17 had maternal diabetes, and 17 were "idiopathic"; however, in one case, a metabolic disorder was found postnatally, and 4 had familiarity for HCM. Outcomes. Ten cases opted for termination of pregnancy. Two cases with DCM and 1 with HCM were lost at follow-up. Out of the cases that continued pregnancy, with known follow-up, mortality was 68.75% in Group 1, 63.6% in Group 2, and 31.3% in Group 3 (the majority with severe ECAs). Surviving cases with DCM and myocarditis improved, 2 with HCM worsened, 6 remained stable, and 26 improved or normalized. Conclusions. Our data show more severe prognosis in DCM and myocarditis and forms with severe associated ECAs.
PMID 20976307
Elevated glucose induces congenital heart defects by altering the expression of tbx5, tbx20, and has2 in developing zebrafish embryos
Liang J, Gui Y, Wang W, Gao S, Li J, Song H. Birth Defects Res A Clin Mol Teratol. 2010 Jun;88(6):480-6.
BACKGROUND: Maternal diabetes increases the risk of congenital heart defects in infants, and hyperglycemia acts as a major teratogen. Multiple steps of cardiac development, including endocardial cushion morphogenesis and development of neural crest cells, are challenged under elevated glucose conditions. However, the direct effect of hyperglycemia on embryo heart organogenesis remains to be investigated.
METHODS: Zebrafish embryos in different stages were exposed to D-glucose for 12 or 24 hr to determine the sensitive window during early heart development. In the subsequent study, 6 hr post-fertilization embryos were treated with either 25 mmol/liter D-glucose or L-glucose for 24 hr. The expression of genes was analyzed by whole-mount in situ hybridization.
RESULTS: The highest incidence of cardiac malformations was found during 6-30 hpf exposure periods. After 24 hr exposure, D-glucose-treated embryos exhibited significant developmental delay and diverse cardiac malformations, but embryos exposed to L-glucose showed no apparent phenotype. Further investigation of the origin of heart defects showed that cardiac looping was affected earliest, while the specification of cardiac progenitors and heart tube assembly were complete. Moreover, the expression patterns of tbx5, tbx20, and has2 were altered in the defective hearts.
CONCLUSIONS: Our data demonstrate that elevated glucose alone induces cardiac defects in zebrafish embryos by altering the expression pattern of tbx5, tbx20, and has2 in the heart. We also show the first evidence that cardiac looping is affected earliest during heart organogenesis. These research results are important for devising preventive and therapeutic strategies aimed at reducing the occurrence of congenital heart defects in diabetic pregnancy.
PMID 20306498
Incidence of congenital heart defects in the Czech Republic--current data
Ceska Gynekol. 2010 May;75(3):221-42.
(Article in Czech)
The study presents current results of analysis of CHD incidences in the Czech Republic in the 1994 - 2008 period. Children born with a CHD make more than 36% out of all children born with a congenital anomaly. CHD themselves represents an important part (more than 40%) of all diagnosed congenital anomalies in the Czech Republic. Over the period of the study there was a slight increase of diagnosed CHD during 1994 - 1999 followed by a slight decrease from 2000 with an exception of 2007 year. The most frequent of diagnosed CHD were ventricular septal defect (Q21.0) and atrial septal defect (Q21.1). Both defects incidences changes influence not only a total CHD but also a total congenital anomalies incidence. An influence of prenatal diagnostics among the five selected CHD was most important in hypoplastic left heart syndrome (Q23.4), less so in others. In prenatal diagnostics group, it is necessary to distinguish between those anomalies, which led to pregnancy termination (parts of both chromosomal and non-chromosomal syndromes and/or association with other severe anomalies) and those in which pregnancy leads to a delivery (late diagnostics, operabile defects, parental decision). CHD can be a part of chromosomal syndromes. In our study, in prenatally diagnosed CHD it was more than 42%. A presence of other associated diagnoses of congenital anomalies in births will significantly influence infant mortality and morbidity.
PMID 20731304
Clinical outcome in Down syndrome patients with congenital heart disease
Cir Cir. 2010 May-Jun;78(3):245-50.
[Article in English, Spanish]
Martínez-Quintana E, Rodríguez-González F, Medina-Gil JM, Agredo-Muñoz J, Nieto-Lago V.
Complejo Hospitalario Universitario Insular-Materno Infantil, Las Palmas de Gran Canaria, Spain. efrenmartinezquintana@yahoo.es Abstract BACKGROUND: Long-term complications of Down syndrome patients with congenital heart disease are poorly known.
METHODS: We carried out a retrospective study of Down syndrome patients with congenital heart disease and patients with atrioventricular septal defect with and without Down syndrome.
RESULTS: Between 2004 and 2008, 317 patients with congenital heart disease were followed-up in the Adult Congenital Heart Disease Unit. Of these patients, 19 (6%) with a mean age of 26.8 +/- 8.1 years had Down syndrome. Atrioventricular septal defect was the most frequent congenital heart disease(63%) followed by ventricular septal defect (26%). Ten patients (53%) were operated on during childhood. Three of these patients required reoperation during adulthood (two patients due to left ventricle outflow tract obstruction and one patient due to left atrioventricular valve insufficiency). Four patients (21%) had Eisenmenger syndrome with improvement of functional class in those treated with bosentan, two patients (10.5%) had bacterial endocarditis and two patients (10.5%) died. No significant differences were seen in left atrioventricular valve insufficiency between atrioventricular septal defect in patients with and without Down syndrome (1.5 +/- 0.9 vs. 1.7 +/- 0.8, p = 0.689).
CONCLUSIONS: Left atrioventricular valve insufficiency and left ventricle outflow tract obstruction were the most frequent long-term complications requiring surgical reintervention in patients with atrioventricular septal defect.
PMID 20642908
2009
Congenital heart disease in 111 225 births in Belgium: birth prevalence, treatment and survival in the 21st century
Acta Paediatr. 2009 Mar;98(3):472-7. Epub 2008 Nov 30.
Moons P, Sluysmans T, De Wolf D, Massin M, Suys B, Benatar A, Gewillig M.
Source
Center for Health Services and Nursing Research, Katholieke Universiteit Leuven, Leuven, Belgium. Philip.Moons@med.kuleuven.be
Abstract AIM: To investigate the birth prevalence, treatment modalities and short-term survival of children with congenital heart disease who were born in 2002. METHODS: We undertook a retrospective review of medical records of all patients who were born in 2002, and were diagnosed, treated and/or followed-up in one of the seven-paediatric cardiology programmes in Belgium. RESULTS: In 111 225 births, 921 children with congenital heart disease were detected, yielding a birth prevalence of 8.3 per 1000. The most frequently occurring conditions were ventricular septal defects (VSDs) (33%), ostium secundum atrial septal defects (18%) and pulmonary valve abnormalities (10%). Thirty-nine percent of the children either had a cardiosurgical operation or catheter intervention. In this study, 4% of the children died. The actuarial survival at 6 months and 1 year of age was 97% and 96%, respectively and remained stable after then. Compared to other heart defects, mortality was higher in univentricular physiology, pulmonary atresia with VSD, left ventricle outflow obstruction and tetralogy of Fallot. CONCLUSION: Survival of congenital heart disease is excellent and continued to improve in the early 21st century. New therapeutic options are increasingly used. This study provides baseline data for the longitudinal follow-up of this cohort.
PMID 19046347
Repair of atrial septal defects on the perfused beating heart
Tex Heart Inst J. 2009;36(5):425-7.
Pendse N, Gupta S, Geelani MA, Minhas HS, Agarwal S, Tomar A, Banerjee A. Source Department of Cardiovascular & Thoracic Surgery, Govind Ballabh Pant Hospital, Delhi University, New Delhi 110002, India. Abstract We present our experience in repairing all varieties of atrial septal defects with the aid of continuous antegrade perfusion of an empty beating heart with normothermic blood. From September 1999 through December 2008, 266 patients (140 females and 126 males; ages 3-53 yr) underwent atrial septal defect closure by this method. Of these patients, 236 had ostium secundum, 21 had sinus venosus, and 9 had ostium primum defects. Three patients also had rheumatic mitral incompetence requiring mitral valve implantation, and 2 also had mitral stenosis requiring valvuloplasty. Preoperative diagnoses were established by 2-dimensional echocardiography and color-flow Doppler study. The size of atrial septal defects ranged from 2 cm through 4.5 cm. Direct repair was performed in 52 patients, and the rest received an autologous pericardial patch. Normothermic perfusion at 4 to 5 mL/(kg.min) kept the heart beating throughout the procedure. All patients survived the procedure with no complication. Twelve patients with ostium secundum atrial septal defect were extubated on the table and discharged within 24 hours of hospitalization. They are categorized as ambulatory cases. All patients remained in sinus rhythm. One patient with a residual shunt required revision of a patch; postoperative echocardiography showed normal left ventricular function and no residual shunt. Total intensive care unit stay was less than 24 hours for all patients.The primary aim of the beating-heart technique is to avoid ischemic-reperfusion injury. It is a safe and effective technique for the closure of all varieties of atrial septal defect.
PMID 19876418
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2763446
2008
Transposition of the great arteries
Orphanet J Rare Dis. 2008 Oct 13;3:27.
Martins P, Castela E. Source Serviço de Cardiologia Pediátrica, Hospital Pediátrico de Coimbra, Coimbra, Portugal. paula_mrtns@yahoo.com
Abstract
Transposition of the great arteries (TGA), also referred to as complete transposition, is a congenital cardiac malformation characterised by atrioventricular concordance and ventriculoarterial (VA) discordance. The incidence is estimated at 1 in 3,500-5,000 live births, with a male-to-female ratio 1.5 to 3.2:1. In 50% of cases, the VA discordance is an isolated finding. In 10% of cases, TGA is associated with noncardiac malformations. The association with other cardiac malformations such as ventricular septal defect (VSD) and left ventricular outflow tract obstruction is frequent and dictates timing and clinical presentation, which consists of cyanosis with or without congestive heart failure. The onset and severity depend on anatomical and functional variants that influence the degree of mixing between the two circulations. If no obstructive lesions are present and there is a large VSD, cyanosis may go undetected and only be perceived during episodes of crying or agitation. In these cases, signs of congestive heart failure prevail. The exact aetiology remains unknown. Some associated risk factors (gestational diabetes mellitus, maternal exposure to rodenticides and herbicides, maternal use of antiepileptic drugs) have been postulated. Mutations in growth differentiation factor-1 gene, the thyroid hormone receptor-associated protein-2 gene and the gene encoding the cryptic protein have been shown implicated in discordant VA connections, but they explain only a small minority of TGA cases.The diagnosis is confirmed by echocardiography, which also provides the morphological details required for future surgical management. Prenatal diagnosis by foetal echocardiography is possible and desirable, as it may improve the early neonatal management and reduce morbidity and mortality. Differential diagnosis includes other causes of central neonatal cyanosis. Palliative treatment with prostaglandin E1 and balloon atrial septostomy are usually required soon after birth. Surgical correction is performed at a later stage. Usually, the Jatene arterial switch operation is the procedure of choice. Whenever this operation is not feasible, adequate alternative surgical approach should be implemented. With the advent of newer and improved surgical techniques and post operative intensive care, the long-term survival is approximately 90% at 15 years of age. However, the exercise performance, cognitive function and quality of life may be impaired.
PMID 18851735
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2577629
http://www.ojrd.com/content/3/1/27
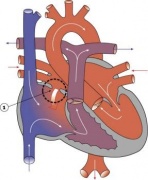
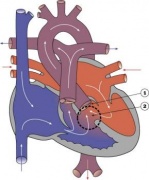
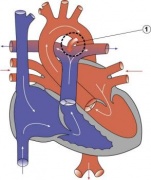
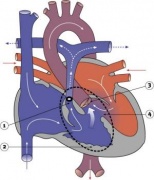
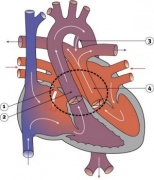
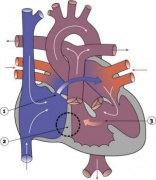
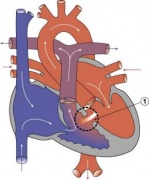
Heart Abnormality Cartoons
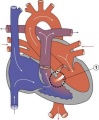
Ventricular Septal Defect
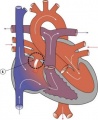
Atrial Septal Defect
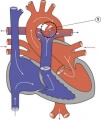
Patent Ductus Arteriosus
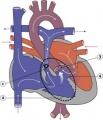
Tetralogy of Fallot
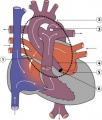
Hypoplastic Left Heart
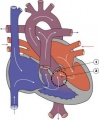
Double Outlet Right Ventricle
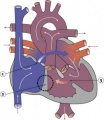
Tricuspid Atresia







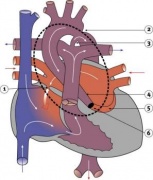
Heart Vessel Abnormality Cartoons
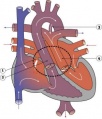
Transposition of the Great Vessels
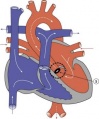
Aortic Stenosis
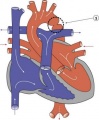
Coarctation of the Aorta
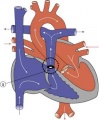
Pulmonary Stenosis
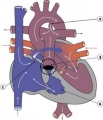
Pulmonary Atresia
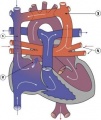
Total Anomalous Pulmonary Venous Connection
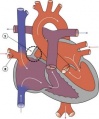
Partial Anomalous Pulmonary Venous Drainage






