Talk:Mitochondria
| About Discussion Pages |
|---|

Glossary Links
Cite this page: Hill, M.A. (2024, April 27) Embryology Mitochondria. Retrieved from https://embryology.med.unsw.edu.au/embryology/index.php/Talk:Mitochondria |
10 Most Recent
Note - This sub-heading shows an automated computer PubMed search using the listed sub-heading term. References appear in this list based upon the date of the actual page viewing. Therefore the list of references do not reflect any editorial selection of material based on content or relevance. In comparison, references listed on the content page and discussion page (under the publication year sub-headings) do include editorial selection based upon relevance and availability. (More? Pubmed Most Recent)
Embryo Mitochondria
<pubmed limit=5>Embryo Mitochondria</pubmed>
Mitochondria
<pubmed limit=5>Mitochondria</pubmed>
2018
Segregation of mitochondrial DNA heteroplasmy through a developmental genetic bottleneck in human embryos
Nat Cell Biol. 2018 Feb;20(2):144-151. doi: 10.1038/s41556-017-0017-8. Epub 2018 Jan 15.
Floros VI1,2, Pyle A3, Dietmann S4, Wei W1,2, Tang WCW5, Irie N5, Payne B3,6, Capalbo A7,8, Noli L9,10, Coxhead J11, Hudson G3, Crosier M12, Strahl H13, Khalaf Y9,10, Saitou M14,15, Ilic D9,10, Surani MA5, Chinnery PF16,17.
Erratum in Author Correction: Segregation of mitochondrial DNA heteroplasmy through a developmental genetic bottleneck in human embryos. [Nat Cell Biol. 2018]
Abstract
Mitochondrial DNA (mtDNA) mutations cause inherited diseases and are implicated in the pathogenesis of common late-onset disorders, but how they arise is not clear1,2. Here we show that mtDNA mutations are present in primordial germ cells (PGCs) within healthy female human embryos. Isolated PGCs have a profound reduction in mtDNA content, with discrete mitochondria containing ~5 mtDNA molecules. Single-cell deep mtDNA sequencing of in vivo human female PGCs showed rare variants reaching higher heteroplasmy levels in late PGCs, consistent with the observed genetic bottleneck. We also saw the signature of selection against non-synonymous protein-coding, tRNA gene and D-loop variants, concomitant with a progressive upregulation of genes involving mtDNA replication and transcription, and linked to a transition from glycolytic to oxidative metabolism. The associated metabolic shift would expose deleterious mutations to selection during early germ cell development, preventing the relentless accumulation of mtDNA mutations in the human population predicted by Muller's ratchet. Mutations escaping this mechanism will show shifts in heteroplasmy levels within one human generation, explaining the extreme phenotypic variation seen in human pedigrees with inherited mtDNA disorders. PMID: 29335530 DOI: 10.1038/s41556-017-0017-8
2017
Dynamic changes in mitochondrial DNA, distribution and activity within cat oocytes during folliculogenesis
Reprod Domest Anim. 2017 Apr;52 Suppl 2:71-76. doi: 10.1111/rda.12851. Epub 2017 Jan 22.
Songsasen N1, Henson LH1, Tipkantha W2,3, Thongkittidilok C1, Henson JH4, Chatdarong K3, Comizzoli P1.
Abstract
Mitochondria play fundamental roles during oocyte development. The accumulation and spatial redistribution of these energy-producing organelles have been linked to the developmental competence of mammalian oocytes. Here, we assessed the copy number, distribution and activity of mitochondria within cat oocytes during folliculogenesis. In Experiment 1, oocytes were recovered from primordial (n = 152), primary (112), secondary (95), early (131), small (118), antral (86) and advanced antral (5) stages follicles, and mitochondria DNA extracted and quantified using qPCR. In Experiment 2, oocytes from pre-antral (n = 44), early antral (n = 66), small antral (n = 59), antral (n = 41) and advanced antral (n = 21) follicles were isolated and stained with CMXRos MitoTracker dye to assess mitochondrial distribution pattern and activity levels. Oocyte's mitochondria DNA (mtDNA) copy numbers gradually increased as folliculogenesis progressed, with a significant shift at the small antral stage (0.5 to <1 mm in diameter). The location of mitochondria gradually shifted from a homogeneous distribution throughout the cytoplasm in pre-antral oocytes to a pericortical concentration in the advanced antral stage. Quantification of CMXRos fluorescent intensity revealed a progressive increase in mitochondrial activity in oocytes from the pre-antral to the large antral follicles. Taken together, these findings demonstrated that cat oocytes undergo dynamic changes in mitochondrial copy number, distribution and activity during folliculogenesis. These significant modifications to this crucial cytoplasmic organelle are likely associated with the acquisition of developmental competency by cat oocytes. KEYWORDS: cat oocyte; follicle development; mitochondria copy number; mitochondrial distribution PMID: 28111812 DOI: 10.1111/rda.12851
Obesity-exposed oocytes accumulate and transmit damaged mitochondria due to an inability to activate mitophagy
Dev Biol. 2017 Jun 1;426(1):126-138. doi: 10.1016/j.ydbio.2017.04.005. Epub 2017 Apr 21.
Boudoures AL1, Saben J1, Drury A1, Scheaffer S1, Modi Z1, Zhang W1, Moley KH2.
Abstract
Mitochondria are the most prominent organelle in the oocyte. Somatic cells maintain a healthy population of mitochondria by degrading damaged mitochondria via mitophagy, a specialized autophagy pathway. However, evidence from previous work investigating the more general macroautophagy pathway in oocytes suggests that mitophagy may not be active in the oocyte. This would leave the vast numbers of mitochondria - poised to be inherited by the offspring - vulnerable to damage. Here we test the hypothesis that inactive mitophagy in the oocyte underlies maternal transmission of dysfunctional mitochondria. To determine whether oocytes can complete mitophagy, we used either CCCP or AntimycinA to depolarize mitochondria and trigger mitophagy. After depolarization, we did not detect co-localization of mitochondria with autophagosomes and mitochondrial DNA copy number remained unchanged, indicating the non-functional mitochondrial population was not removed. To investigate the impact of an absence of mitophagy in oocytes with damaged mitochondria on offspring mitochondrial function, we utilized in vitro fertilization of high fat high sugar (HF/HS)-exposed oocytes, which have lower mitochondrial membrane potential and damaged mitochondria. Here, we demonstrate that blastocysts generated from HF/HS oocytes have decreased mitochondrial membrane potential, lower metabolites involved in ATP generation, and accumulation of PINK1, a mitophagy marker protein. This mitochondrial phenotype in the blastocyst mirrors the phenotype we show in HF/HS exposed oocytes. Taken together, these data suggest that the mechanisms governing oocyte mitophagy are fundamentally distinct from those governing somatic cell mitophagy and that the absence of mitophagy in the setting of HF/HS exposure contributes to the oocyte-to-blastocyst transmission of dysfunctional mitochondria. PMID: 28438607 DOI: 10.1016/j.ydbio.2017.04.005 [Indexed for MEDLINE]
2014
The Mitochondrial Aminoacyl tRNA Synthetases: Genes and Syndromes
Int J Cell Biol. 2014;2014:787956. Epub 2014 Feb 4.
Diodato D, Ghezzi D, Tiranti V. Author information
Abstract
Mitochondrial respiratory chain (RC) disorders are a group of genetically and clinically heterogeneous diseases. This is because protein components of the RC are encoded by both mitochondrial and nuclear genomes and are essential in all cells. In addition, the biogenesis and maintenance of mitochondria, including mitochondrial DNA (mtDNA) replication, transcription, and translation, require nuclear-encoded genes. In the past decade, a growing number of syndromes associated with dysfunction of mtDNA translation have been reported. This paper reviews the current knowledge of mutations affecting mitochondrial aminoacyl tRNAs synthetases and their role in the pathogenic mechanisms underlying the different clinical presentations. PMID 24639874
Dual roles for ubiquitination in the processing of sperm organelles after fertilization
BMC Dev Biol. 2014 Feb 15;14(1):6. [Epub ahead of print]
Hajjar C, Sampuda KM, Boyd L.
Abstract
BACKGROUND: The process of fertilization involves a cell fusion event between the sperm and oocyte. Although sperm contain mitochondria when they fuse with the oocyte, paternal mitochondrial genomes do not persist in offspring and, thus, mitochondrial inheritance is maternal in most animals. Recent evidence suggests that paternal mitochondria may be eliminated via autophagy after fertilization. In C. elegans, sperm-specific organelles called membraneous organelles (MO) cluster together with paternal mitochondria immediately after fertilization. These MOs but not the mitochondria become polyubiquitinated and associated with proteasomes. The current model for the elimination of paternal mitochondria in C. elegans is that ubiquitination of the MOs induces the formation of autophagosomes which also capture the mitochondria and cause their degradation. RESULTS: Sperm-derived mitochondria and MOs show a sharp decrease in number during the time between sperm-oocyte fusion and the onset of mitosis. During this time, paternal mitochondria remain closely clustered with the MOs. Two types of polyubiquitin chains are observed on the MOs: K48-linked ubiquitin chains which are known to lead to proteasomal degradation and K63-linked ubiquitin chains which have been linked to autophagy. K48-linked ubiquitin chains and proteasomes show up on MOs very soon after sperm-oocyte fusion. These are present on MOs for only a short period of time. Maternal proteasomes localize to MOs and sperm proteasomes localize to structures that are at the periphery of the MO cluster suggesting that these two proteasome populations may have different roles in degrading paternal material. K63-linked ubiquitin chains appear on MOs early and remain throughout the first several cell divisions. CONCLUSIONS: Since there are two different types of polyubiquitin chains associated with sperm organelles and their timing differs, it suggests that ubiquitin has two or more roles in the processing of sperm components after fertilization. The K63 chains potentially provide a signal for autophagy of paternal organelles, whereas the K48 chains and proteasomes may be involved in degradation of specific proteins.
PMID 24528894
http://www.biomedcentral.com/1471-213X/14/6
2013
Unique insights into maternal mitochondrial inheritance in mice
Proc Natl Acad Sci U S A. 2013 Aug 6;110(32):13038-43. doi: 10.1073/pnas.1303231110. Epub 2013 Jul 22.
Luo SM1, Ge ZJ, Wang ZW, Jiang ZZ, Wang ZB, Ouyang YC, Hou Y, Schatten H, Sun QY. Author information
Abstract In animals, mtDNA is always transmitted through the female and this is termed "maternal inheritance." Recently, autophagy was reported to be involved in maternal inheritance by elimination of paternal mitochondria and mtDNA in Caenorhabditis elegans; moreover, by immunofluorescence, P62 and LC3 proteins were also found to colocalize to sperm mitochondria after fertilization in mice. Thus, it has been speculated that autophagy may be an evolutionary conserved mechanism for paternal mitochondrial elimination. However, by using two transgenic mouse strains, one bearing GFP-labeled autophagosomes and the other bearing red fluorescent protein-labeled mitochondria, we demonstrated that autophagy did not participate in the postfertilization elimination of sperm mitochondria in mice. Although P62 and LC3 proteins congregated to sperm mitochondria immediately after fertilization, sperm mitochondria were not engulfed and ultimately degraded in lysosomes until P62 and LC3 proteins disengaged from sperm mitochondria. Instead, sperm mitochondria unevenly distributed in blastomeres during cleavage and persisted in several cells until the morula stages. Furthermore, by using single sperm mtDNA PCR, we observed that most motile sperm that had reached the oviduct for fertilization had eliminated their mtDNA, leaving only vacuolar mitochondria. However, if sperm with remaining mtDNA entered the zygote, mtDNA was not eliminated and could be detected in newborn mice. Based on these results, we conclude that, in mice, maternal inheritance of mtDNA is not an active process of sperm mitochondrial and mtDNA elimination achieved through autophagy in early embryos, but may be a passive process as a result of prefertilization sperm mtDNA elimination and uneven mitochondrial distribution in embryos. KEYWORDS: assisted reproductive technologies, mitophagy, paternal inheritance
PMID 23878233
Recurrent tissue-specific mtDNA mutations are common in humans
PLoS Genet. 2013 Nov;9(11):e1003929. doi: 10.1371/journal.pgen.1003929. Epub 2013 Nov 7.
Samuels DC1, Li C, Li B, Song Z, Torstenson E, Boyd Clay H, Rokas A, Thornton-Wells TA, Moore JH, Hughes TM, Hoffman RD, Haines JL, Murdock DG, Mortlock DP, Williams SM.
Author information
Abstract Mitochondrial DNA (mtDNA) variation can affect phenotypic variation; therefore, knowing its distribution within and among individuals is of importance to understanding many human diseases. Intra-individual mtDNA variation (heteroplasmy) has been generally assumed to be random. We used massively parallel sequencing to assess heteroplasmy across ten tissues and demonstrate that in unrelated individuals there are tissue-specific, recurrent mutations. Certain tissues, notably kidney, liver and skeletal muscle, displayed the identical recurrent mutations that were undetectable in other tissues in the same individuals. Using RFLP analyses we validated one of the tissue-specific mutations in the two sequenced individuals and replicated the patterns in two additional individuals. These recurrent mutations all occur within or in very close proximity to sites that regulate mtDNA replication, strongly implying that these variations alter the replication dynamics of the mutated mtDNA genome. These recurrent variants are all independent of each other and do not occur in the mtDNA coding regions. The most parsimonious explanation of the data is that these frequently repeated mutations experience tissue-specific positive selection, probably through replication advantage.
Comment in Genomics: Organ roles for mitochondrial mutations? [Nat Rev Genet. 2014]
PMID 24244193
2012
Effect of Maternal Age on the Ratio of Cleavage and Mitochondrial DNA Copy Number in Early Developmental Stage Bovine Embryos
J Reprod Dev. 2012 Dec 26. [Epub ahead of print]
Takeo S, Goto H, Kuwayama T, Monji Y, Iwata H. Source Tokyo University of Agriculture, Kanagawa 243-0034, Japan. Abstract Age-associated deterioration in both the quality and quantity of mitochondria occurs in older women. The main aim of this study was to examine the effect of age on mitochondrial DNA copy number (mtDNA number) in early developmental stage bovine embryos as well as the dynamics of mtDNA number during early embryo development. Real-time PCR was used to determine mtDNA number. In vitro-produced embryos 48 h after insemination derived from Japanese black cows, ranging in age from 25 to 209 months were categorized based on their cleavage status. There was an overall negative relationship between the age of the cow and cleavage status, to the extent that the ratio of embryos cleaved over the 4-cell stage was greater in younger cows. The mtDNA number did not differ among the cleaved status of embryos. In the next experiment, oocytes collected from each donor cow were divided into 2 groups containing 10 oocytes each, in order to compare the mtDNA number of mature oocytes and early developmental stage embryos within individuals. Upon comparing the mtDNA number between oocytes at the M2 stage and early developmental stage 48 h post insemination, mtDNA number was found to decrease in most cows, but was found to increase in some cows. In conclusion, age affects the cleaving ability of oocytes, and very old cows (> 180 months) tend to have lower mtDNA numbers in their oocytes. The change in mtDNA number during early development varied among individual cows, although overall, it showed a tendency to decrease.
PMID 23269452
Heteroplasmy of mouse mtDNA is genetically unstable and results in altered behavior and cognition
Cell. 2012 Oct 12;151(2):333-43. doi: 10.1016/j.cell.2012.09.004.
Sharpley MS1, Marciniak C, Eckel-Mahan K, McManus M, Crimi M, Waymire K, Lin CS, Masubuchi S, Friend N, Koike M, Chalkia D, MacGregor G, Sassone-Corsi P, Wallace DC. Author information
Abstract Maternal inheritance of mtDNA is the rule in most animals, but the reasons for this pattern remain unclear. To investigate the consequence of overriding uniparental inheritance, we generated mice containing an admixture (heteroplasmy) of NZB and 129S6 mtDNAs in the presence of a congenic C57BL/6J nuclear background. Analysis of the segregation of the two mtDNAs across subsequent maternal generations revealed that proportion of NZB mtDNA was preferentially reduced. Ultimately, this segregation process produced NZB-129 heteroplasmic mice and their NZB or 129 mtDNA homoplasmic counterparts. Phenotypic comparison of these three mtDNA lines demonstrated that the NZB-129 heteroplasmic mice, but neither homoplasmic counterpart, had reduced activity, food intake, respiratory exchange ratio; accentuated stress response; and cognitive impairment. Therefore, admixture of two normal but different mouse mtDNAs can be genetically unstable and can produce adverse physiological effects, factors that may explain the advantage of uniparental inheritance of mtDNA. Copyright © 2012 Elsevier Inc. All rights reserved. Comment in The problem with mixing mitochondria. [Cell. 2012] Chromosome biology: mixing it up. [Nat Rev Mol Cell Biol. 2012]
PMID 23063123
2011
Postfertilization autophagy of sperm organelles prevents paternal mitochondrial DNA transmission
Science. 2011 Nov 25;334(6059):1144-7. doi: 10.1126/science.1211878. Epub 2011 Oct 27.
Al Rawi S1, Louvet-Vallée S, Djeddi A, Sachse M, Culetto E, Hajjar C, Boyd L, Legouis R, Galy V. Author information
Abstract
In sexual reproduction of most animals, the spermatozoon provides DNA and centrioles, together with some cytoplasm and organelles, to the oocyte that is being fertilized. Paternal mitochondria and their genomes are generally eliminated in the embryo by an unknown degradation mechanism. We show that, upon fertilization, a Caenorhabditis elegans spermatozoon triggers the recruitment of autophagosomes within minutes and subsequent paternal mitochondria degradation. Whereas the nematode-specific sperm membranous organelles are ubiquitinated before autophagosome formation, the mitochondria are not. The degradation of both paternal structures and mitochondrial DNA requires an LC3-dependent autophagy. Analysis of fertilized mouse embryos shows the localization of autophagy markers, which suggests that this autophagy event is evolutionarily conserved to prevent both the transmission of paternal mitochondrial DNA to the offspring and the establishment of heteroplasmy. Comment in Development: autophagy eliminates paternal mitochondria. [Nat Rev Mol Cell Biol. 2011] Re: Postfertilization autophagy of the sperm organelles prevents paternal mitochondrial DNA transmission. [Eur Urol. 2012] Development. Inheriting maternal mtDNA. [Science. 2011]
PMID 22033522
Cell Biology lecture
http://php.med.unsw.edu.au/cellbiology/index.php?title=Cell_Mitochondria
This lecture introduces the cytoplasmic organelles that produce the energy required for cellular processes to occur. In recent years mitochondria have also been shown to have important roles in other cellular functions, in particular, cell death by apoptosis. This second role will be covered in detail in later lectures in this current series.
File:Mitochondria fl.jpg File:08lungtem.jpg
Mitochondrion is singular, Mitochondria is plural
Objectives
- Broad understanding of processes requiring energy within the cell
- Brief understanding structure and function of plant chloroplast
- Understand the structure and function of plant and animal mitochondria
- Brief understanding of mitochondria evolution
- Brief understanding of mitochondrial abnormalities
Double Membrane Organelles
- Nucleus - all eukaryotes
- Chloroplasts - plants
- Mitochondria - plants and animals
History Mitochodria
1857 Kölliker discovers mitochondria in muscle
1929 Karl Lohmann discovered ATP
1940s and 1950s ATP is formed in cell respiration in mitochondria and photosynthesis in chloroplasts of plants
1960 Efraim Racker and co-workers isolated, from mitochondria, the enzyme "F o F 1 ATPase" now call ATP synthase
1963 There’s DNA in those organelles DNA is directly visualized in first chloroplasts and then mitochondria, from the JCB Archive.
1992 Wallace identified degenerative disease caused by mtDNA mutations
1997 Nobel Prize in Chemistry - The three laureates have performed pioneering work on enzymes that participate in the conversion of the "high-energy" compound adenosine triphosphate (ATP).
- Paul D. Boyer and John E. Walker "for their elucidation of the enzymatic mechanism underlying the synthesis of adenosine triphosphate (ATP)"
- Jens C. Skou "for the first discovery of an ion-transporting enzyme, Na+, K+ -ATPase
Evolution Mitochondria
- primitive Eubacterium
- symbiotic relationship with eukaryotic cell
- circular DNA
- see antibiotic-induced deafness due to similarity of mitochondrial and bacterial ribosomes
- genes transferred to nucleus
- mitochondrial genome bp
- 366,924 Arabidopsis
- 16,569 Human
- 5966 Plasmodium
Chloroplasts
- Plant cell structure.png
Plant cell structure
- Chloroplast1.jpg
Plant Chloroplast organelles
- Double membrane cytoplasmic organelle
- present in photosynthetic Eubacteria, algae and plants
- thought to originate as an endosymbiotic cyanobacteria (blue-green algae)
Function
- photosynthesis
- chlorophyll captures light energy
- chloroplasts interact with peroxisomes
Structure
- flat discs usually 2 to 10 micrometer in diameter and 1 micrometer thick.
- plants 5 μm in diameter and 2.3 μm thick
- inner and an outer phospholipid membrane
- intermembranous space
- stroma
- stacks of thylakoids (site of photosynthesis)
- contains copies of small circular DNA
- ribosomes
- proteins transported to the chloroplast
(MH - will not cover this cell organelle in any depth in current course)
Mitochondria
- Greek, mito = thread; chondrion = granule
- Located throughout cytoplasmic compartment
- has itself several membrane enclosed compartments
- each compartment has different function
- Ancient aerobic organisms in symbiosis (endosymbiosis)
- present in all cells
Mitochondria Function
- Energy production
- Respiratory chain
- Signaling
- Apoptosis role
- Programmed cell death
Mitochondria Structure
- Double membrane
- outer membrane
- intermembrane space
- inner membrane
- crista (plural, cristae)
- originally considered specialized folds of the inner membrane
- variable invaginations with narrow tubular connections to each other and by crista junctions to the peripheral region of inner membrane
- matrix
Mitochondria Shape
- Come in different shapes & sizes
- Can rapidly change shape (minutes)
EM: Mitochondria EM: Mitochondria EM: Mitochondria Mitochondrial Morphology
Mitochondria Location
- cells with high energy requirements: Muscle, sperm tail, flagella
- generally located where energy consumption is highest in the cell
- Mitochondria (fibroblasts)
- Mitochondria (sperm)
- Packed around initial segment
- Energy for sperm motility, microtubules (9+2)
Mitochondria Components
Outer Membrane
- porin - membrane channel, allows ions and metabolites into the mitochondria (<5000 daltons)
Intermembrane Space
- similar to the cytosol with respect to the small molecules it contains
- also enzymes that use ATP
Inner Membrane
- cardiolipin - phospholipid, makes membrane impermeable to ions
- transport proteins - permeable to molecules required in the matrix
Cristae
- increase inner membrane surface area
- tubular, vesicular or flat cristae
- Adenosine triphosphate (ATP) synthase
- respiratory electron transfer chain proteins
- transport proteins
Matrix
- metabolic enzymes of citric acid cycle (=Krebs) (100s of enzymes) (MH- do not need to know biochemical details of this cycle)
- genetic material DNA, tRNA, ribosomes
Mitochondria DNA

- double stranded circular DNA (mitoDNA. mtDNA)
- 1981 complete human sequence (16,569 nucleotides)
- 37 genes
- encodes 13 polypeptides involved in oxidative phosphorylation
- remaining genes transfer RNA (tRNA) and ribosomal RNA (rRNA)
- multiple copies within the matrix
- maternally inherited
- remainder encoded by nuclear DNA
- proteins made in cytosol and imported into mitochondria
Links: Home Reference - Mitochondrial DNA
Mitochondria Protein Synthesis
- yeast - Petite mutants
- all mitochondrion-encoded gene products missing
- forms small anaerobic colonies
- organelle is constructed entirely from nucleus-encoded proteins
Many mitochondrial proteins are encoded by nuclear DNA
- synthesis begins in the cell cytoplasm
- imported into the mitochondria
- targeting similar to signal sequence for RER
- once in matrix signal sequence is cleaved (by Hsp70)
- protein then folds (by Hsp60)
- proteins for mitochondrial membrane or intermembranous space
- additional signal following matrix localization
Mitochondrial targeting signal (MTS) - alternating amino acid pattern (amphipathic helix) with a few hydrophobic amino acids and a few plus-charged amino acids at the N terminus.
Links: Replication and preferential inheritance of hypersuppressive petite mitochondrial DNA | Home Reference - Mitochondrial DNA
Mitochondrial Division and Fusion
Mitochondria Fission
- Mitochondrial Division
- Divide independently of the whole cell cycle
- Generated by existing mitochondria
- inward furrowing like bacterial division
- mitochondria lack FtsZ ring (seen in bacteria)
- rely on dynamin on the cytosolic face for fission
Mitochondrial inheritance
Mitochondrial Fusion
- when two separate mitochondria join as one
- fission and fusion considered to be balanced
- disruption causes normal tubular network of mitochondria to fragment into short rods or spheres
- Requires large GTPases
- Mitofusins 1 and 2 (Mfn1, Mfn2) and OPA1
- Mfn1 and Mfn2 for outer membrane fusion
- OPA1 for inner membrane fusion
Links: Preventing Mitochondrial Fission Impairs Mitochondrial Function and Leads to Loss of Mitochondrial DNA | Mitochondrial dynamics–fusion, fission, movement, and mitophagy–in neurodegenerative diseases]
Energy Production
- Electrochemical proton gradient across the inner mitochondrial membrane is used to drive ATP synthesis MBoC - The general mechanism of oxidative phosphorylation
Respiration
- Raw Materials
- Oxygen
- Pyruvate & Fatty Acids
- Products
- Carbon Dioxide
- Adenosine Triphosphate (ATP)
Apoptosis
Mitochondria in addition to energy production, have a second major function related to programmed cell death by apoptosis.
- cytochrome C release activates caspases
- other changes include
- electron transport, loss of mitochondrial transmembrane potential
- altered cellular oxidation-reduction
- Bcl-2 family proteins (pro- and antiapoptotic)
- Vesicular Mitochondria
- begin to appear during the release of cytochrome C which initiates mitochondrial mediated apoptosis
- transformation from normal morphology
- with an inner boundary membrane connected to lamellar cristae via crista junctions
- multiple vesicular matrix compartments
- facilitates membrane fission or fragmentation as the matrix is fragmented at this stage
- fragmentation of the mitochondrion requires only outer membrane fission
(MH- this topic will be covered again in the Cell Death Lecture)
Links: Lecture - Cell Death 1 | Lecture - Cell Death 2 | Movie - Vesicular Mitochondria | Role of mitochondria in apoptosis | Ibioseminar - Apoptosis Part 2: Factors Involved in the Intrinsic Pathway of Apoptosis (27:39 minutes)
Methods
Dynabead Isolation
- Mitochondria isolation using magnetic beads conjugated to antibodies to mitochondrial surface markers.
MitoTracker Probes
- MitoTracker probes are cell-permeant mitochondrion-selective dyes.
Links: Bovine pulmonary artery endothelial cells (BPAEC) - LysoTracker Red and MitoTracker Green | Bovine pulmonary artery endothelial cell - probes to mitochondria (red), peroxisomes (green), and the nucleus (blue) | Molecular Probes - Probes for Mitochondria | Spectral characteristics of the MitoTracker probes |
Apoptosis Assays
- Cytochrome c Releasing Apoptosis Assays - detection of cytochrome c translocation from mitochondria into cytosol during apoptosis. Commercial Kit PDF
- Cytochrome c normally located in the space between the inner and outer mitochondrial membranes
- Mitochondrial transmembrane potential changes (JC-1, cationic dye) Commercial Kit 1 | Commercial Kit 2
- Healthy cells - accumulates and aggregates in the mitocondria, bright red fluorescence.
- Apoptotic cells - altered mitochondrial transmembrane potential causes dye to remain in the cytoplasm in monomer form, green fluorescence.
MitoChip
- Microarray of both strands of the entire human mitochondrial coding sequence (15,451 bp).
- detecting germ line and heteroplasmic mutations by complete mitochondrial genome resequencing Affymetrix Commercial Microarray Manual
- oligonucleotide probes synthesized using standard photolithography and solid-phase synthesis, and is able to sequence >29 kb of double-stranded DNA in a single assay.
The Human MitoChip: a high-throughput sequencing microarray for mitochondrial mutation detection. Maitra A, Cohen Y, Gillespie SE, Mambo E, Fukushima N, Hoque MO, Shah N, Goggins M, Califano J, Sidransky D, Chakravarti A. Genome Res. 2004 May;14(5):812-9. PMID: 15123581 | http://genome.cshlp.org/content/14/5/812.abstract Genome Res. 2004]
Abnormalities
Nuclear transfer of mitochondrial DNA
- mitochondria to the nucleus generates nuclear copies of mitochondrial DNA (numts)
- Integration can appear as neutral polymorphism or associated with human diseases (insertion of mtDNA into genes,5 known cases), the mitochondrial genome remains intact in the individuals.
- insertion at the breakpoint junction of a reciprocal translocation between chromosome 9 and 11.
- insertion into splice site mutation in the human gene for plasma factor VII (causes severe plasma factor VII deficiency, bleeding disease)
- insertion into exon 14 of the GLI3 gene causes a premature stop codon (associated with Chernobyl)
- insertion into exon 2 of MCOLN1, eliminated proper splicing of the gene (mucolipidosis IV).
- insertion in exon 9 of the USH1C gene (Usher syndrome type IC)
Above examples from: PMID: 15361937
Mitochondrial Myopathies
- A group of several diseases
- Kearns-Sayre syndrome (KSS) Genes and Disease - Kearns-Sayre syndrome
- Leigh's syndrome Genes and Disease - Leigh's syndrome
- mitochondrial DNA depletion syndrome (MDS)
- mitochondrial encephalomyopathy, lactic acidosis and strokelike episodes (MELAS) Genes and Disease - MELAS
- myoclonus epilepsy with ragged red fibers (MERRF)
- mitochondrial neurogastrointestinal encephalomyopathy (MNGIE)
- neuropathy, ataxia and retinitis pigmentosa (NARP) Genes and Disease - NARP
- Pearson syndrome Genes and Disease - Pearson syndrome
- Progressive external ophthalmoplegia (PEO) Genes and Disease - Progressive External Ophthalmoplegia
Friedreich's ataxia (FRDA)
- mutation in gene for frataxin (a mitochondrial iron (Fe) chaperone)
- mechanism may be a mitochondrial iron (Fe) loading and reactive oxygen species
- animal models show H2O2 is an important pathogenic substrate underlying the phenotypes arising from frataxin deficiency
- Genes and Diseases - Friedreich's ataxia
Charcot–Marie–Tooth type 2A
- inherited neuropathy
- caused by mutations in mitofusin 2
- proper regulation of mitochondrial dynamics required in neurons
Leber Hereditary Optic Neuropathy
- maternally inherited cause of blindness Genes and Diseases - LHOM
- mutation of mitochondrial DNA (mtDNA)
- three common mtDNA mutations: G11778A, T14484C, G3460A
Aminoglycoside-induced deafness
- due to aminoglycoside antibiotic treatment Genes and Diseases - Aminoglycoside-induced deafness
- gentamycin, streptomycin, and tobramcyin
- similarity of mitochondrial ribosomes to bacterial ribosomes
Other
Adenoid Cystic Carcinoma (ACC)
Mitochondrial mutations in adenoid cystic carcinoma of the salivary glands. Mithani SK, Shao C, Tan M, Smith IM, Califano JA, El-Naggar AK, Ha PK. PLoS One. 2009 Dec 30;4(12):e8493. PMID: 20041111
- "Mitochondrial mutation is frequent in salivary ACCs. The high incidence of amino acid changing mutations implicates alterations in aerobic respiration in ACC carcinogenesis. D-loop mutations are of unclear significance, but may be associated with alterations in transcription or replication."
Cyclic Vomiting Syndrome?
Links: Genes and Diseases -Mitochondria and Disease | GeneReviews - Mitochondrial Disorders Overview | Genes and Diseases - Friedreich's ataxia | Muscular Dystrophy Association - Mitochondrial Myopathies | The Cleveland Clinic - Mitochondrial Myopathies | Genes and Diseases | OMIM - Online Mendelian Inheritance in Man
References
Textbooks
Essential Cell Biology
- Chapter 13 Energy Generation in Mitochondria and Chloroplasts
- Chapter 14 Intracellular Compartments and Transport
Molecular Biology of the Cell
Alberts, Bruce; Johnson, Alexander; Lewis, Julian; Raff, Martin; Roberts, Keith; Walter, Peter New York and London: Garland Science; c2002
Molecular Cell Biology
Lodish, Harvey; Berk, Arnold; Zipursky, S. Lawrence; Matsudaira, Paul; Baltimore, David; Darnell, James E. New York: W. H. Freeman & Co.; c1999
- Mitochondria Are the Principal Sites of ATP Production in Aerobic Cells
- Figure 16-7. A three-dimensional diagram of a mitochondrion cut longitudinally
The Cell- A Molecular Approach
Cooper, Geoffrey M. Sunderland (MA): Sinauer Associates, Inc.; c2000
- The Cell - A Molecular Approach - III. Cell Structure and Function 10. Bioenergetics and Metabolism - Mitochondria, Chloroplasts, and Peroxisomes
- Mitochondria
Search Online Textbooks
- "mitochondria" Molecular Biology of the Cell | Molecular Cell Biology | The Cell- A molecular Approach | Bookshelf
- "chloroplast" Molecular Biology of the Cell | Molecular Cell Biology | The Cell- A molecular Approach | Bookshelf
Books
PubMed
- PubMed is a service of the U.S. National Library of Medicine that includes over 18 million citations from MEDLINE and other life science journals for biomedical articles back to 1948. PubMed includes links to full text articles and other related resources. PubMed
- PubMed Central (PMC) is a free digital archive of biomedical and life sciences journal literature at the U.S. National Institutes of Health (NIH) in the National Library of Medicine (NLM) allowing all users free access to the material in PubMed Central. PMC
- Online Mendelian Inheritance in Man (OMIM) is a comprehensive compendium of human genes and genetic phenotypes. The full-text, referenced overviews in OMIM contain information on all known mendelian disorders and over 12,000 genes. OMIM
- Entrez is the integrated, text-based search and retrieval system used at NCBI for the major databases, including PubMed, Nucleotide and Protein Sequences, Protein Structures, Complete Genomes, Taxonomy, and others Entrez
Search Pubmed
- "mitochondria" PubMed reviews | PubMed all articles | PMC reviews | PMC all articles | OMIM | Entrez all databases
Reviews
- Mitochondrial fragmentation in neurodegeneration. Knott AB, Perkins G, Schwarzenbacher R, Bossy-Wetzel E. Nat Rev Neurosci. 2008 Jul;9(7):505-18. Review. PMID: 18568013
- Emerging functions of mammalian mitochondrial fusion and fission. Chen H, Chan DC. Hum Mol Genet. 2005 Oct 15;14 Spec No. 2:R283-9. Review. PMID: 16244327
Articles
- Bcl-x L increases mitochondrial fission, fusion, and biomass in neurons. Berman SB, Chen YB, Qi B, McCaffery JM, Rucker EB 3rd, Goebbels S, Nave KA, Arnold BA, Jonas EA, Pineda FJ, Hardwick JM. J Cell Biol. 2009 Mar 9;184(5):707-19. Epub 2009 Mar 2. PMID: 19255249
Mitochondrion Images
For a full selection of see Cell Biology Images - Mitochondria Images
- 07lungtem.jpg
cell from lung (tem)
- 08lungtem.jpg
cell from lung (tem)
- Mitochondria 1 tem.jpg
Mitochondria (tem)
- 02lungtem.jpg
cell from lung (tem)
- Cardiac muscle mitochondria.jpg
Cardiac muscle mitochondria (tem)
- Heart mitochondria.jpg
Heart mitochondria (tem)
- Heart papillary muscle.jpeg
Heart_papillary_muscle (tem)
- Mitochondria location mitosis.jpg
Morphological changes in the cellular mitochondrial network during mitosis
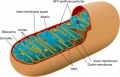
Mitochondrion structure cartoon
- Mitochondrial membraneous compartments.jpg
Mitochondrial membraneous compartments
- Mitochondrion rat liver.jpg
Mitochondrion rat liver
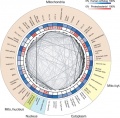
Eukaryotic mitochondrial genomes
- Mitochondrial ROS production cartoon.jpg
Mitochondrial ROS production
- Mitochondrial oxidative phosphorylation pathway.jpg
Mitochondrial genome and the oxidative phosphorylation pathway
- Apoptosis pathway cartoon.jpg
Apoptosis pathway cartoon
- Models of Mitochondrial Membrane Permeabilization.jpg
Models of Mitochondrial Membrane Permeabilization
- Viral Proteins Mitochondrial Membrane Permeabilization.jpg
Viral Proteins Mitochondrial Membrane Permeabilization
- Bcl-XL Functions Like a Dominant-Negative Bax.jpg
Bcl-XL Functions Like a Dominant-Negative Bax

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Movies
- Mitochondrial replication factories DNA replication factories in mitochondria divide even in the absence of mitochondrial DNA, according to Meeusen and Nunnari.
- Apoptotic cells show a transient loss in mitochondrial membrane potential Waterhouse et al. suggest that the transient loss of mitochondrial membrane potential that can be seen in individual apoptotic cells may be masked by cell-to-cell asynchrony when looking at a population of cells.
- Mitochondria slow down for calcium Mitochondria move along microtubules, but Yi et al. find that they slow down when calcium levels rise, probably so that they can help buffer the ion back to normal levels.