2012 Group Project 6

Hearing Development
Introduction
CAN YOU HEAR ME! – Hearing is one of the most important senses and an inherent part of human life. It widens our scope of life and allows us to work, relax, communicate, learn and form memories. The sense of hearing has evolved through the years in vertebrates and is inherent for both hunting and surviving. The sound energy produced has to be converted into an electrical signal for us to process this in our brain. For successful transmission the correct development of the ear is of utmost important. Not only the formation of the structures but also their correct assembly is imperative to normal ear functioning. In this project we will discuss the development of the human ear from implantation to birth by shining light on the differentiation of cells and signalling mechanisms that lead to normal development. We will then talk about some abnormal processes and mutations which lead to various structural and functional diseases. Technologies to detect and overcome these abnormalities will also be considered.
History
| Date | Description |
| 1536 | The first clear distinction is made between the middle and inner ear. How to observe the middle ear ossicles is also explained. [1] [2] Nicola Massa. |
| 1543 | De fabrica by Andreas Vesalius was published which identified and named the incus and the malleus. Also identified was the tensor tympani and the anatomical position of the oval window to the round window [3]. Andreas Vesalius |
| 1561 | Observationes anatomicae is published and describes the tympanic membrane. The stapes ossicle is also discovered and named. [4] Gabriele Falloppio |
| 1563 | In the book De Auditus Organis, it was proposed that the tympanic membrane is connected to the nasopharynx. This tube was named after the writer of the book. He also completed the diescritpion of the tensor tymapani: [5]Bartolomeo Eustachi |
| 17th Century | The Metal ear was one of the first hearing aids created. It was placed over the ear to channel sound into the ear. |
| 1648 | Athanasius Kircher describes the ear trumpet: Athanasius Kircher |
| 1683 | The bony labyrinth is described in more detail. It is determined the spiral lamina has a bony and membranous part, and it divides the spiral duct into an upper and lower compartment. [2]Duverney. |
| 1704 | The book De aure humana tractatus with information regarding the anatomy, physiology and pathology of the ear is published.[6] Valsalva. |
| 1707 | It is proposed that the labyrinth contains fluid instead of air. [6] Valsalva. |
| 1789 | The membranous labyrinth is shown to consist of semicircular canals and the vestibular sacs. These form one system and are different to the periosteum. [7] Scarpa. |
| 1831 | It was discovered that the membranous labyrinth develops from a pit in the skin.It was also discovered that the spiral lamina is hollow. [8] Huschke. |
| 1851 | The cochlea is discovered in the inner ear, and parts of it, including the sensory epithelium, the spiral ganglion, the tectorial membrane, and the stria vascularis are described. Alfonso Corti. |
| 1898 | The first electric hearing aid, the Akouphone, was developed. A carbon transmitter allowed the hearing aid to be portable. Miller Reese Hutchison. |
| 1961 | Georg Von Békésy was awarded the Nobel Prize in Physiology or Medicine. He was awarded on this research on the sound waves at different frequencies on the nerve fibres. the research showed that the hair cells were activated along the cochlea correlated to the position of the cochlea. Georg Von Békésy. |
| 1978 | For the first time, a cochlear implant allowed a person to hear again. Professor Graeme Clark. |
| 2012 | For the first time, human embryonic stem cells were used and proven to be effective in restoring hearing. See Current Research below and the news article. |
Adult Ear: Overview of Anatomy and Physiology

In order for mammals to hear, sound energy in the air has to move the hair cells located inside the inner ears of mammals. The ear provides us with the suitable apparatus to convert the longitudinal sound waves into mechanical ones in order to move the hair cells located in the inner ear.[9] The Pinna or the auricle is designed such that the sound waves coming in get directed into the external auditory meatus (EAM). The waves travel through the EAM hit the tympanic membrane or eardrum, which causes it to vibrate.[9] The malleus, which is in contact with the tympanic membrane, moves which in turn moves the other ossicles. The ossicles namely malleus, incus and stapes provide solid medium for the waves to travel through and act as a lever increasing the amplitude of the sound wave 20 times.[9] [10]

The oval window marks the boundary between the middle and the inner ear, which constitutes of an outer bony labyrinth and an inner membranous labyrinth.[10] The cochlea, which is a coil resembling a snail’s shell, houses the organ of Corti, the peripheral receptor for hearing. Histologically the cochlea is divided into 3 parts scala vestibule, scala media and the scala tympani divided by the vestibular and basilar membrane respectively. [9]
The sound waves are transmitted from the stapes into the oval window. The vibrations cause movement of the perilymph fluid in the scala vestibuli into the scala media causing movement of basilar membrane and bending of the hair cells.[10] The bending of the cells causes them to activate which sends signals down the vestibulocochlear nerve.[9] The nerve impulses travel from this neve through the brainstem to the thalamus to the primary auditory cortex, which is located on the transverse temporal gyrus. [9] [11]
The basilar membrane has a tonotopic organisation and human ears can hear sounds ranging from 20-20000 Hz. Near the apex of the cochlea the basilar membrane is wide and loose whereas near the base is narrow and tight hence enabling us to hear different frequencies.[9]
Development

The development of the outer and middle ear is attributed to the pharyngeal arches one and two. All the germ layer namely endoderm, mesoderm and ectoderm contribute to its formation. [12] The entire inner ear, as well as the neurons which innervate the sensory organ, are derived from the otic placode. [13]
The pharyngeal arches arise as a series of bulges arising laterally from the embryo head around 3-4 weeks of human development. The arches have a consistent organisation of the endoderm, ectoderm and mesoderm.[10] The ectoderm forms the outer surface of the arch with the core made up of mesoderm. Next to the mesoderm core on the opposite the ectoderm is the endoderm. All three layers contribute to the formation of outer and middle ear structures. In between the arches the ectoderm and endoderm come in contact with each other forming a continuous sheath on either side of the mesoderm, forming groves externally and arches internally. Inside the mesoderm core of each arch lies a specific cell population that goes onto develop into a nerve, cartilage and artery. [14] The otic placode is a thickened portion of ectoderm located on the each side of the developing head of the embryo, next to the hindbrain.[13] This will form an otic vesicle (otocyst) and further develop into the structures of the inner ear.
We will consider the outer, middle and inner ear separately.
Outer Ear
Pinna

Post development the anatomy of the external ear simply consists of the pinna or the auricle and external acoustic/auditory meatus. This forms as follows: [10] [13]
- The auricle develops around the first and second pharyngeal arches as a series of auricular enlargements or hillocks around the 5th week of development.
- Gradually by the 6th week the hillocks grow in size and increases to six in number, three on the first arch and 3 on the second arch, the first of which starts at the bottom anterior side, then following with the others in a clockwise direction.
- In the 7th week the hillocks enlarge further and each of the arches contributes to a specific part of the pinna. The first arch gives rise to the tragus, helix and cymba concha whereas arch 2 gives rise to concha, antihelix and antitragus. The differentiation starts around the neck region but as the mandible develops the pinna structures move more cranially.
- It is not until the 12th week that the fusion of these various parts occur and is completed by week 20th.
Various genetic markers and coordinated signalling mechanisms are required for normal development of the pinna.[10] One of which is the EYA1 gene, which is imperative for formation of the pinna.[15] Mice with a homologous Eya1 null gene either have malformed or absent ears. Since Eya1 gene plays a role in the formation of cartilage, its absence doesn’t allow the ear mesenchyme to convert into ear cartilage. [16] Another gene important for pinna cartilage formation is the Bmp5 gene that is expressed later in development; mutations cause malformation of the perichondrium and thus cartilage formation.[17] Another gene important for early patterning of the pinna is the Hox2 gene.[14] Mutations will lead to the formation of a shapeless protuberance rather than a normal shaped pinna. Hox2 gene is expressed in the second pharyngeal arch and the malformations in the ear are contributed to the parts derived from the second pharyngeal arch like the antitragus, antihelix and concha. [16] [18]
External Auditory Meatus
The External Auditory Meatus (EAM) is derived from ectoderm that forms the first pharyngeal groove, situated between the 1st and the 2nd pharyngeal arches. It forms as follows: [10]
- At 8 weeks of gestation a c-shaped skeletal structure develops from ectoderm of the first pharyngeal cleft migrating and meeting the mesoderm of the first grove called the tympanic ring that controls and coordinates the invagination of the ectoderm. The tympanic ring is a transient embryological structure, which eventually gets integrated into the temporal bone and provides anchorage to the eardrum. Medial to the ring lays the endoderm of the first pharyngeal arch that contributes to the formation of the tympanic or middle ear cavity.
- At 12 weeks of gestation the tympanic ring begins to ossify in a sequential fashion via endomembranous ossification forming the bony part of the EAM. The ring starts to condense at the proximal end of the 1st pharyngeal arch, goes around the circumference of the cleft and invades the 2nd arch.
- Starting at the tip of the 1st pharyngeal cleft the ectodermal cells begin to proliferate filling the lumen of the meatus forming a meatal plug which medially extends in a disc like fashion during week 10. The proliferation follows the path of the tympanic ring with the mesoderm tissue between the ring and the meatus forming a fibrous layer.
- In week 13 of development the innermost part of the meatal plug makes a contact with malleus.
- This innermost part of the disc splits in week 15, leaving a thin endoderm layer behind which contributes to formation of the tympanic membrane.
- By the middle of 16th week the meatus although full in length is still narrow and curved and it is not until week 18 that it gets fully expanded and complete.
Since the correct development of EAM is heavily dependent on the tympanic ring, any mutation in the tympanic ring causes the abnormal growth of the EAM.[19] Many genes are required to coordinate and control tympanic ring formation most prominent being the Gsc and Prx1 genes. Chimeric studies have revealed that tympanic ring in Gsc null mice fails to ossify whereas the ring whereas mice with null Prx1 gene have an over ossified tympanic ring.[20] In both cases the meatus fails to form indicating a delicate balance between the expressions of both genes is required for proper ring development hence correct formation of EAM.[20] Likewise for pinna formation Hox2 gene is also essential for differentiation of the pharyngeal arch into the tympanic ring and the formation of EAM.[19] [16]
Middle Ear
Tympanic Membrane
Tympanic membrane or the eardrum forms the boundary between the outer and middle ear and responsible for transmitting the soundwaves coming in from the EAM to the mechanical waves going into the ossicles.[16] During 8th week of gestation the development of tympanic membrane begins when the first funnel shaped ectodermal cleft meets the endodermal pouch with the mesenchyme growing in-between. This mesenchyme eventually becomes thinner and forms the eardrum during week 12. The diameter of the membrane grows three fold between week 11 and 16 and the membrane fuses with the tympanic ring. [19]

The 1st pharyngeal cleft (ectoderm) gives rise to the EAM and the tympanic membrane comes from the mesoderm from the 1st pharyngeal arch. [21]The 1st pharyngeal pouch (endodermal in origin) proliferates to giving rise to the tubotympanic recess at 3rd gestational week.[22] During week 7 the 2nd pharyngeal arch constricts the tympanic recess dividing it into a medial portion that form the Eustachian tube and a lateral portion that forms the tympanic cavity. [19]
The Ossicles
The middle ear constitutes of the skeletal structures called ossicles, which amplify and transmit sound to the inner ear.
The middle ear consists of the middle ear cavity and it are housed the 3 ossicles – the malleus, incus and stapes from lateral to medial. The mesenchymal tissue or the neural crest cells from 1st and 2nd pharyngeal arch contributes to the formation of the ossicular chain. [23]
Two main theories underlie the formation of the ossicles. First is the classical theory according to which the malleus and incus develops from the first pharyngeal arch whereas stapes develops from the 2nd pharyngeal arch. [23] Another theory proposes that malleal head and body of incus comes from the first pharyngeal arch while the second arch gives rise to stapes, handle of malleus and long process of incus. [23]

Inner Ear
The entire inner ear, as well as the neurons which innervate the sensory organ, are derived from the otic placode.
The otic placode is a thickened portion of ectoderm located on the each side of the developing head of the embryo, next to the hindbrain.[13]It is generally visible after gastrulation, once the first 5 to 10 pairs of somites have formed - week 3 of human embryonic development. Invagination occurs next, which creates the otocyst. Patterning occurs and as a result, we can see the start of formation of the cochlea during week 5. The otocyst will develop into the different components of the inner ear: the cochlea, the semicircular canals with cristae, the utricle, the saccule and the vestibulo-acoustic ganglion. [13] Chondrification and ossification of the otic capsule takes places from week 9 and week 11 respectively.

The Otic Placode
Induction of the otic placode
Experiments with molecular markers have revealed that several steps are needed for induction of the otic placode.
We will briefly consider the three major steps:
1 Pre-placodal domain
The pre-placodal domain is a narrow strip of the ectoderm adjacent to the anterior neural plate after gastrulation. Different placodes arise from the pre-placodal domain. All the craniofacial sensory organs, including the ear, develop from these different placodes located at the periphery of the neural plate.
Various evidence indicates the existence of this pre-placodal region:
- Morphology indicates a thickened band of ectoderm around the anterior neural plate in some species, including mice and humans. As time progresses, this thickening will only be present at the locations where the different craniofacial placodes differentiate - including the otic placode.[24]
- Experiments have also shown that the placodes will only develop in the correct location, if rotation of the ectoderm along the anteroposterior axis takes place at the open neural plate stage. If rotation takes place at a later time, the placodes will form at incorrect places.[25]
- Gene expression has also indicated that particular genes are present in the pre-placodal domain. These genes belong to the Dlx, Six, Eya, Iro, BMP, Foxi and Msx families. Glavic et al. (2004) has shown that 'loss and gain of function of some of these genes resulted in the widening or reduction of the pre-placodal field'. Linked to this was also the domain of expression of some placode-specific genes; which either enlarged or diminished.[26]

2 Pre-otic field
Once the general placodal state has been established, the identity of each placode is induced by local signals. The optic placode is induced by various signals, including Pax8, Pax2, Fibroblast Growth Factors (FGFs), and many transciption factors.
In particular the FGFs are significant otic inducers. Signalling occurs from various rhombomeres from the hindbrain and the cranial paraxial mesoderm located beneath the area of the otic placode. For example, in mice FGF3 is expressed in rhombomeres 5 and 6, whereas FGF10 is expressed in the underlying mesoderm.[27] [28] Mutations of FGF3 and FGF10 have been investigated. Results showed that mice with a mutation of either FGF3 or FGF10 developed an abnormal otic vesicle, and a combination of the two mutants resulted in failure to form an otic vesicle.[28]

3 Otic placode/epidermis fate decision
In the presence of FGF signalling, Wnt signalling can significantly influence the next step, which is the otic placode/epidermis fate decision. According to the review article by Ohyama et al. (2007) ‘Cells receiving high levels of Wnt signalling differentiate as otic placode, while cells receiving little or no Wnt signalling differentiate as epidermis.’[13]
The first evidence regarding the contribution of Wnt signalling came from experiments with the otic ectoderm of chicks.
- Data showed that specific marker genes, such as Pax2, were induced to a greater extend with FGF19 and Wnt8c present as compared to FGF19 alone [29]. Ladher et al.(2000) hypothesised that FGF19 induced Wnt8c, and together they induced the otic gene markers.
Another possibility is the independent action of FGFs and Wnt signalling.
- Studies have shown that Wnt signalling onto Pax2+ cells results in differentiation of those cells into otic placode tissue. Pax2+ cells that were not exposed to Wnt signalling differentiate as epidermis [30].
- Wnt signalling also suppresses Foxi2, resulting in a Foxi2-negative area of particular size, which then allows for FGFs to induce otic genes [30]. In this case, Wnt signalling determined the size of the otic placode, yet acted independently from FGF signalling.
- Phillips et al. (2004) studied the role of FGF and Wnt signalling; specifically looking at FGF3, FGF8 and Wnt8. Their data showed that Wnt8 is not absolutely necessary for otic induction, however it is required for timely initiation of the otic field. [31]
The neural domain

During the early stages of embryonic development, a neural competent domain is established. This domain will eventually give rise to neurons and hair cells. Various signalling pathways are required to initially create this domain and to maintain it later on. [32]
- FGF and Sox
FGF signalling and the Sox genes are essential for the establishment of the neural competent domain. An important aspect of Sox genes is that they have a state of self-renewal, as well as a state of neural commitment. [33] As investigated by Rex et al. (1997) and Pevny and Placzek (2005), "SoxB1 genes (Sox1, Sox2, Sox3) have been linked directly to ectodermal cells that are competent to acquire neural fate, and the commitment of cells to a neural fate".[34] [35]
Before the otic placode becomes distinct, Sox3 is already expressed in a broad area around the location of the future otic placode. Later in it will only be found in the proneural region of the otic placode. Based on this, the review by Alsina et al. (2009) suggests that a neural fate acquisition occurs prior to otic placode formation.[32] During the early stages of development of the otocyst, Sox2 and Sox3 are found in proliferating cells within the proneural region. Both are expressed when neurons are generated, however, Sox3 switches off and only Sox2 remains during further development of the ear. This suggests that the ongoing expression of Sox2 plays a role in sensory cell development, as explained in the review by Alsina et al (2009).[32]
- Notch signalling
Notch signalling is required for several developmental processes, including the maintenance of the neural competent domain.
In the notch signalling pathway, notch is the receptor, with most of its ligands being transmembrane proteins. Signalling is therefore restricted to neighbouring cells. Research by Daudet and Lewis (2005) reveiled how notch signalling plays a role in inner ear development, including:
- Notch signalling mediates lateral inhibition and thereby controls the differentiation of hair cells and supporting cells - in particular in the vestibular regions [36]
- An early phase of Notch activity promotes formation of prosensory patches [36]
- Other signalling pathways are likely to cooperate with Notch to specify prosensory regions of the otocyst [36]
The Otocyst

Patterning of the otocyst
The otocyst, also known as the otic vesicle, is present once invagination of the otic placode has occurred. Regionalisation of the otocyst results in the topological organisation of the ear. The neural tube affects the patterning of the otocyst, and FGF, Wnt and Hh signalling pathways are also known to play a role. FGF and Wnt rely on signals from the hindbrain. [37]

Prosensory patches emerge within the otocyst. These develop as a differentiate from the neural competent domain.[38]
- haircells (sensory patch)
- neurons (neural competent domain)
As proposed in recent models by Neves et al. (2011): Jagged 1 functions through lateral induction to activate Notch signalling. Notch signalling then functions through lateral inhibition and regulates Sox2 expression. Sox2 specifies sensory fate within the prosensory domains. "This confines sensory competence to the prosensory patches, ensuring the development of sensory organs of the correct size and location." [38]
Establishing polarity

The different axis of the inner ear are fixed at different points in time. This means different signals are involved to establish the polarity and allow for development.[39] The dorsal-ventral polarity is very significant in the development of inner ear structures. The ventral inner ear consists of the cochlea and saccule, and the dorsal inner ear is made up of semicircular canals, endolymphatic duct, cristae and utricle.
The neural tube plays a role in patterning of the placode and it has been shown similar signals from the neural tube are also needed to establish the dorso-ventricular axis of the inner ear.[40]
- Shh signalling
The notochord produces Shh, which helps in patterning the dorso-ventricular axis of the neural tube. [41] This signal diffuses further and also affects the developing otocyst, where a gradient of Shh receptors is located from the dorsal to ventral aspect. [42] Dorso-ventricular patterning and development of inner ear structures – in particular ventral structures – is achieved by the graded response to Shh.
As reviewed by Grooves and Fekete (2012) “data suggests that Shh acts on the ventral otocyst directly to regulate cochlear development, and that dorsal development can be regulated by signals from tissues adjacent to the otocyst that require Shh signaling for their normal development.”[43]
- Wnt signalling
Shh signalling for the ventral aspect is complemented by signals for the dorsal aspect of the inner ear. Wnt signalling takes places in the otic placode, as described above. This is initially as a gradient from medial to lateral [44], which later in the otocyst will be a gradient from doral to ventral [45]
As further suggested in the review by Grooves and Fekete (2012) “Wnt and Shh signals regulate different inner ear genes in different ways, with opposing gradients of Shh and Wnt signaling regulating the spatial localization of transcription factors, ultimately leading to the differentiation of a correctly patterned inner ear.” [43]
- Hedgehog signalling

It is also crucial for normal development of inner ear structures that Hedgehog signalling is repressed. [46]
Formation of inner ear structures
Further development of the inner ear occurs as the otic vesicle differentiates into the membranous labyrinth. This is a continuous structure, surrounded by an otic capsule. The otic capsule is mesoderm derived from the base of the skull and will later chondrify, holding the organs of balance and hearing, as well as the cranial nerve associated with them. [47]
1 Semi-circular canals
As reviewed by Carey and Amin (2006), the semi-circular canals - the organ of balance, also known as the vestibular apparatus - develops from the superior surface of the otocyst. This occurs as the cranial end elongates as 3 little expansions, with the remainder forming the utricle.[48]

2 Cochlea
The cochlea - the organ of hearing - develops from the inferior surface of the otocyst, as reviewed by Fritzsch et al. (2011). [49] This occurs as the caudal end elongates and curves 2.5 times (in humans), forming the cochlear duct; the remainder forms the saccule.[49] Growth of the cochlear duct is controlled by the growth of the organ of Corti, which itself depends upon hair cell development [50]. At first, a simple epithelium is still present within the cochlear duct, however, at a later stage we will see the distinct differentiation of epithelium into stereocilia hair cells, the organ of Corti and the saccular macula.[49] [51]
3 Vestibulocochlear nerve (CN VIII)
During its development, this cranial nerve receives contribution from the otocyst and cranial neural crest cells. It contains bipolar neurons, vestibular neurons and cochlear neurons.[52] It exits through the internal acoustic meatus (IAM) to conduct signals to the brain for processing.
Summary inner ear
| SUMMARY OF THE INNER EAR DEVELOPMENT |
1. Pre-placodal domain: After gastrulation, a pre-placodal domain is present adjacent to the anterior neural plate. The various placodes will arise from this domain. 2. Pre-otic field: Local signals establish the identity of each placode, including the otic placode. FGF signals from both rhombomeres and the cranial paraxial mesoderm are important otic inducers. 3. Otic placode/epidermis fate decision: signalling occurs to determine precisely which cells become the otic placode and which cells aquire the epidermal fate. FGF and Wnt signalling is necessary in this step, which could possibly be dependent or independent of each other.
|
Abnormal Hearing
In this section we will discuss with examples the different types of congenital abnormalities of the ear leading to hearing impairment or deafness. Apart from the Genetic section (whereby there are 5 ways the genes can be transferred), we will provide 3 examples in each due to the number of causes of congenital deafness. Note that the conditions can be split into syndromic and non syndromic along with conductive hearing loss and sensorineural hearing loss.
Genetic
| Genetic abnormality | Description | Image |
| Mutation of GJB2 gene |
|
|
| Autosomal dominant hearing loss |
|
|
| Autosomal recessive hearing loss |
|
|
| X linked hearing loss |
|
|
| Mitochondrial hearing loss |
|




Genetic Syndromes
| Syndrome | Description | Image |
| Usher Syndrome |
|
|
| Pendred Syndrome |
|
|
| Goldenhar Syndrome |
|
|



Environmental
Infections
| Organism | Description |
| Toxoplasmosis |
|
| Rubella |
|
| Cytomegalovirus |
|
Drugs
| Drug | Description |
| Alcohol consumption during pregnancy |
|
| Isotretinoin |
|
| Thalidomide |
|
Structural malformations of the ear
| Structural malformation | Description | Image |
| Stenosis |
|
|
| Enlarged vestibular aqueduct |
|
|
| Microtia |
|
|



Technologies to detect
The importance of having a neonatal screening test for hearing within the early days of life are important for the rest of that individual’s life. Screening is used to identify the children most at risk of having a congenital hearing problem. The importance of having an early diagnosis and having a method of intervention is crucial for the development of speech and language and learning of the child later in life, as reviewed by Oudesluys-Murphy ‘’et al’’. 1996. [89] Screening tests allow for the detection of hearing loss within the few days of life. This could be due to a dysfuctional cochlea or various other problems as discussed in our abnormal hearing section. Within Australia there are a few simple test that allow newborns to be tested fast, accurately and reliable [89].
Oto-acoustic testing

The oto-acoustic test measures the integrity of the inner ear. This involves the cochlea and its physiological effects - the production of an otoacoustic emission in response to sound [90]. It involves inserting a probe into the ear canal, which then produces clicks or tones that are normally picked up by the cochlea. The function and healthiness of the cochlea can be measured, as the different parts of the cochlea respond to different forms of stimuli in terms on pitch and frequency [90].
For this test, there are two methods used for screening the hearing in newborns.
• The production of a single click or tone called the Transient Evoked Otoacoustic Emission test (TEOAE) [90]
• The production of two simultaneous tones named the Distortion Product Otoacoustic Emissions Test (DPOAE) [90]
Auditory Brainstem Response
The Auditory Brainstem Response tests the neurological function of the auditory brainstem. It is more so used as a referral test, rather than a screening test, as generally hearing loss is already suspected [91]. This test uses a click or a tone, which causes the production of impulses by the neurons from the auditory nerve, travelling along the auditory pathway. This is then measured and detected by external electrodes that are on the scalp and the earlobe of the newborn. [91]
Automated Brainstem Response
This cheaper and quicker method uses an ear cup that is fitted over the infant’s ear [92]. This then sends out a stimulus and a response is measured through a series of electrodes that are connected to the earlobes, scalp, shoulders and neck[92]. The automated brainstem response uses a computer to visualise and calculate the results, and produce a result of either ‘pass/refer’. Data of the patient are cross-referenced with a collection of results regarded as normal responses[92].
OtoSCOPE
A new genetic testing method called OtoSCOPE discovered in 2010 has greatly increased the efficiency for testing nonsyndromic hearing loss and Usher syndrome. It provides a great diagnostic tool, as all the genes related to hearing loss can be sequenced simultaneously thereby decreasing cost and time. The patients can be informed relatively early for any genetic abnormalities therefore providing early prognosis and genetic counselling. [93]
Technologies to overcome the problems
Depending on the hearing loss, there are different types of technologies to overcome this. Types of hearing lost are conductive and sensori-neutral . One type is the conductive, caused by the damage or blockage of the outer or middle ear, the other is sensori-neutral, where the damage is to the auditory pathway or nerve. In some cases there is a combination of the two. And there are two current technologies that overcome this problem and they are the the hearing aid and the cochlea ear implant.
Hearing aid

Hearing Aids are a major advance in helping people hearing, they work by amplifying the sounds so they can be detected even by the damaged inner ear. This method of hearing relies on the healthiness or still relative functioning cochlea. The damage of the sensori-neural pathway is overcome by the amplification of sounds to compensate the loss of hearing cells or hair cells in the cochlea. This is proportionate to the amount of hair cells still functioning. This amplification of the sounds, helps the patient hear and listen and communicate with others. This however also amplifies background noise. They consists of the microphone, amplifier and a speaker. From the sound being received by the microphone, this converted by the microphone into electrical impulses and replays it to the amplifier. The amplifier sends the impulses though the speaker into the ear as an increased sound. [94]
Cochlea Ear Implant
Cochlea ear implants help the hearing impaired to listen. This is through having a device to provide a sense of sound. The cochlea implants consists of a few major components in helping the patient hear. It consists of a microphone, which detects the environmental sounds, a small electronic processor that selects the sound produced from the microphone. There is also a transmitter and the stimulator. This converts the signal that comes off the electronic processor into electrical impulses, which leads up the final component which is the electrode array. This picks up the electrical impulses and delivers them to the correct region of the auditory nerve. These methods of hearing or sensing sound are used only in severer or profoundly hearing impaired who have a dysfunctioning cochlea. [95]

Current Research
- Hair cell differentiation
A recently published paper by Pan et al. (2012) investigated the various levels and durations of expression of a particular transcription factor, which is necessary for hair cell differentiation. The Atoh1 transcription factor was tested for by using conditional knock-out mice - Atoh1-cre [96]. They determined that reduced levels of Atoh1 resulted in the progressive loss of hair cells from the organ of Corti shortly after birth [96]. Similar data was obtained when the deletion of the Atoh1 transcription factor was delayed. Slight differences were noted between inner and outer hair cells: the loss of inner hair cells was more significant than the loss of outer hair cells. This indicates that Atoh1 may play a role in the differential development of inner and outer hair cells.


- Stem cell therapy
The discovery of stem cells has led to an entirely new era in research, and the potential to prevent and treat conditions such as hearing loss. It has been discovered that stem cells are present in the inner ear. In non-mammalian vertebrates, these stem cells allow the inner ear sensory epithelium to recover and function again after being damaged. This is not the case when it comes to mammals. Cells capable of regeneration are found in the vestibular sensory epithelium and in the neonatal cochlea. [97] However, only very limited regeneration may take place after damage has been done to the inner ear. Research is ongoing in various laboratories worldwide, such as at the University of Sheffield, as to how stem cells can be used to regain the sense of hearing. Early September 2012 there was an incredible breakthrough, as scientists from the university used human embryonic stem cells to restore hearing in a common form of deafness. This proves human stem cells could be used to repair the damaged ear and will hopefully lead to various other stem-cell based therapies.

- Middle ear homeostasis
A recent article by Morris et al. (2012) investigated the mechanisms responsible for the ion homeostasis in the middle ear and how this relates to disease. They used BALB/c mice for immunohistochemistry of particular ion homeostasis factors of the middle and inner ear [98]. This was then used to compare transport and barrier mechanisms and identify what is present in the tympanic cavity. Next, the middle ears received transtympanic injections with heat-killed Haemophilus influenza to determine if these channels are impacted by inflammation [98]. Data shows that cellular hypertrophy occurred and localization of ion channels, such as aquaporins, was preserved within the inflamed middle ear epithelium[98]. Morris et al. suggested that these channels could be used as a therapeutic target.
- Molecular mechanisms - ongoing project
The HEARing Cooperative Research Centre (CRC) is involved in many ongoing research projects. Professor Doug Hilton supervises one of the projects: Genomic & molecular therapeutic approaches to environmental and age-related hearing loss.
A major public health issue is presbycusis - age-related hearing loss. It is known that apoptosis of cells within the cochlea plays a role; however, the many other factors involved and their molecular mechanisms are not yet fully understood. This research project is looking at the genes, proteins and regulatory pathways involved in both hearing and hearing-loss. The aim is to use this knowledge to identify molecules that can be targeted to prevent and/or treat hearing loss.
Glossary
- Caudal: towards the embryonic tail
- Chondrification: the process that results in the formation of cartilage
- Cranial: towards the head of the embryo
- Cristae: the sensory organ of rotation located in the semicircular canal of the inner ear
- Ectoderm: the outermost layer of the three primary germ cell layers in the very early embryo
- Epidermis: surface epithelium of the skin, superficial to the dermis
- FGF – Fibroblast Growth Factor: a family of polypeptides that are involved in embryonic development and function as growth and differentiation factors.
- Foxi2: forkhead box I2 gene. Located on chromosome 10, position 26.2, this gene produces a transcription factor protein, which regulates the activity of other genes
- Gastrulation: The inward migration of cells
- Hindbrain: The lower part of the brainstem, comprising the cerebellum, pons, and medulla oblongata
- Incus: the second ossicle of the middle ear, located between the malleus and stapes
- Inferior: towards the bottom
- Invagination: the infolding of tissue, such as the otic placode
- Lateral inhibition: process whereby one cell takes on a state and the adjacent cells takes on the opposite state
- Malleus: the first ossicle of the middle ear, located between the tympanic membrane and the incus
- Mesoderm: the middle layer of the three primary germ cell layers in the very early embryo
- Neural plate: a thickened plate of ectoderm along the dorsal midline of the early vertebrate embryo that gives rise to the neural tube and crests
- Neural tube: A hollow structure formed after gastrulation, from which the brain and spinal cord form
- Nonsyndromic deafness: Hearing impairment not affiliated with other signs or symptoms
- Notch: this receptor allows for binding of particular ligands and hence enables signalling between neighbouring cells.
- Open neural plate stage: stage of the neural plate before closure into the neural tube
- Ossification: the process that results in the formation of bone
- Otic placode: a thickening of the ectoderm on the outer surface of a developing embryo from which the ear develops
- Otocyst – otic vesicle: The structure formed by invagination of the embryonic ectodermal tissue that develops into the inner ear
- Paraxial mesoderm: The mesoderm located alongside the neural tube
- Pax2: the paired box 2 gene. Located on chromosome 10, position 24, this gene produces a transcription factor protein, which regulates the activity of other genes
- Pre-placodal domain: An ectodermal domain with multipotential progenitors that contribute to sense organs and cranial sensory ganglia
- Proneural region – neural competent domain: a region of the otic placode involved in neurogenesis
- Prosensory region: region containing a population of cells that can develop into hair cells or or supporting cells
- Rhombomere: a segment of the developing rhombencephalon (hindbrain segment)
- Saccule: one of two otolith organs. The smaller of the two fluid-filled cavities forming part of the labyrinth of the inner ear
- Somite: A segmental mass of mesoderm in the vertebrate embryo, occurring in pairs along the notochord
- Sox1: SRY (sex determining region Y)-box 1. Located on chromosome 13, position 32, this gene produces a transcription factor protein and is mainly expressed in the developing central nervous system.
- Sox2: SRY (sex determining region Y)-box 2. Located on chromosome 3, position 26.33, this gene produces a transcription factor protein. It is needed for embryonic stem cell pluripotency and neural stem cell self-renewal.
- Sox3: SRY (sex determining region Y)-box 3. Located on chromosome 3, position 26.33, this gene produces a transcription factor protein. Needed to maintain undifferentiated neural cells, formation of the hypothalamo-pituitary axis and sex differentiation.
- Stapes: the third ossicle of the middle ear, located between the incus and the oval window
- Superior: towards the top
- Syndromic deafness: Hearing impairment occurring with additional abnormalities in the body
- Tonotopic organisation: the structural arrangement which allows for sounds of different frequencies to be detected and processed separately
- Topological organisation: the organisation of an area, such as the otocyst, according to the structures it relates to, such as the semicircular canals and the cochlea.
- Utricle: one of two otolith organs. The larger of the two fluid-filled cavities forming part of the labyrinth of the inner ear
- Vestibular region: the region of the inner ear close to the cochlea. Here the semicircular canals converge
- Vestibulo-acoustic ganglion: The cranial ganglion of cranial nerve 8
- Wnt signaling molecules: a highly conserved family of proteins that control interactions between cells
Please note, to ensure the accuracy of the descriptions of terms above, various dictionaries were used, including the medical dictionary and the Britannica online encyclopedia.
References
- ↑ <pubmed>4598483</pubmed>
- ↑ 2.0 2.1 <pubmed>6341584</pubmed>
- ↑ <pubmed>22581496</pubmed>
- ↑ <pubmed>22965774</pubmed>
- ↑ <pubmed>7029021</pubmed>
- ↑ 6.0 6.1 <pubmed>PMC1142106</pubmed>
- ↑ <pubmed>11314703</pubmed>
- ↑ <pubmed>1910378</pubmed>
- ↑ 9.0 9.1 9.2 9.3 9.4 9.5 9.6 <pubmed>16269359</pubmed>
- ↑ 10.0 10.1 10.2 10.3 10.4 10.5 10.6 <pubmed>11237469</pubmed>
- ↑ <pubmed>21774850</pubmed>
- ↑ <pubmed>8287791</pubmed>
- ↑ 13.0 13.1 13.2 13.3 13.4 13.5 <pubmed>17891709</pubmed>
- ↑ 14.0 14.1 <pubmed>11698185</pubmed>
- ↑ <pubmed>9853969</pubmed>
- ↑ 16.0 16.1 16.2 16.3 <pubmed>14674478</pubmed>
- ↑ <pubmed>7958439</pubmed>
- ↑ <pubmed>17104502</pubmed>
- ↑ 19.0 19.1 19.2 19.3 <pubmed>10976045</pubmed>
- ↑ 20.0 20.1 <pubmed>1441991</pubmed>
- ↑ <pubmed>17346562</pubmed>
- ↑ <pubmed>2921547</pubmed>
- ↑ 23.0 23.1 23.2 <pubmed>18803631</pubmed>
- ↑ <pubmed>15531360</pubmed>
- ↑ <pubmed>14100031</pubmed>
- ↑ <pubmed>15242793</pubmed>
- ↑ <pubmed>7789270</pubmed>
- ↑ 28.0 28.1 <pubmed>12810586</pubmed>
- ↑ <pubmed>11110663</pubmed>
- ↑ 30.0 30.1 <pubmed>16452098</pubmed>
- ↑ <pubmed>14757644</pubmed>
- ↑ 32.0 32.1 32.2 <pubmed>19247974</pubmed>
- ↑ <pubmed>15863505</pubmed>
- ↑ <pubmed>9215646</pubmed>
- ↑ <pubmed>15721738</pubmed>
- ↑ 36.0 36.1 36.2 <pubmed>15634704</pubmed>
- ↑ <pubmed>17891710</pubmed>
- ↑ 38.0 38.1 <pubmed>21266409</pubmed>
- ↑ <pubmed>9389659</pubmed>
- ↑ <pubmed>16325169 </pubmed>
- ↑ <pubmed>18621990</pubmed>
- ↑ <pubmed>12231626</pubmed>
- ↑ 43.0 43.1 <pubmed>22186725</pubmed>
- ↑ <pubmed>20171206 </pubmed>
- ↑ <pubmed>16452098</pubmed>
- ↑ <pubmed>20223756</pubmed>
- ↑ <pubmed>22778034</pubmed>
- ↑ <pubmed>16552774</pubmed>
- ↑ 49.0 49.1 49.2 <pubmed>21256948</pubmed>
- ↑ <pubmed>18579736</pubmed>
- ↑ <pubmed>16145671</pubmed>
- ↑ <pubmed>12530227</pubmed>
- ↑ 53.0 53.1 <pubmed>10980526</pubmed>
- ↑ <pubmed> 11216656 </pubmed>
- ↑ <pubmed> 14979964 </pubmed>
- ↑ <pubmed> 1163535 </pubmed>
- ↑ <pubmed>PMC3335728</pubmed>
- ↑ <pubmed> 16434480 </pubmed>
- ↑ <pubmed> 16452831 </pubmed>
- ↑ <pubmed>3776519</pubmed>
- ↑ <pubmed> 18285825 </pubmed>
- ↑ <pubmed>10829494 </pubmed>
- ↑ <pubmed> 9408975 </pubmed>
- ↑ 64.0 64.1 <pubmed>3041362</pubmed>
- ↑ <pubmed>16311017</pubmed>
- ↑ <pubmed>12454967</pubmed>
- ↑ <pubmed> 10356003 </pubmed>
- ↑ <pubmed> 9988811 </pubmed>
- ↑ <pubmed>84910</pubmed>
- ↑ <pubmed>5949097</pubmed>
- ↑ <pubmed>2817948</pubmed>
- ↑ <pubmed>11017784</pubmed>
- ↑ <pubmed>9792846</pubmed>
- ↑ <pubmed>19111256</pubmed>
- ↑ <pubmed>3067168</pubmed>
- ↑ <pubmed>22519989</pubmed>
- ↑ <pubmed>16192480</pubmed>
- ↑ <pubmed>19766534</pubmed>
- ↑ 79.0 79.1 <pubmed>9161611</pubmed>
- ↑ <pubmed>12795509</pubmed>
- ↑ <pubmed>PMC1859978</pubmed>
- ↑ <pubmed>1440418</pubmed>
- ↑ <pubmed>21507989</pubmed>
- ↑ <pubmed> 9892864 </pubmed>
- ↑ <pubmed> 16570074 </pubmed>
- ↑ <pubmed>7877418</pubmed>
- ↑ <pubmed>12671419</pubmed>
- ↑ <pubmed> 10890145 </pubmed>
- ↑ 89.0 89.1 <pubmed>8789756</pubmed>
- ↑ 90.0 90.1 90.2 90.3 <pubmed>8220282</pubmed>
- ↑ 91.0 91.1 <pubmed>11667937</pubmed>
- ↑ 92.0 92.1 92.2 <pubmed>11343042</pubmed>
- ↑ <pubmed>21078986</pubmed>
- ↑ National Institute of Health, Hearing Aid Basics, http://www.nidcd.nih.gov/health/hearing/pages/hearingaid.aspx, Access date September 19, 2012
- ↑ <pubmed>6028666</pubmed>
- ↑ 96.0 96.1 <pubmed>22279587</pubmed>
- ↑ <pubmed>8456285</pubmed>
- ↑ 98.0 98.1 98.2 <pubmed>22720014</pubmed>
External Links
Study: how mitochondrial DNA defects cause inherited deafness
Study: Sound-induced length changes in outer hair cell stereocilia
Video: The process of hearing and how it works
Video: Deaf toddler finally hears his mom's voice
External Links Notice - The dynamic nature of the internet may mean that some of these listed links may no longer function. If the link no longer works search the web with the link text or name. Links to any external commercial sites are provided for information purposes only and should never be considered an endorsement. UNSW Embryology is provided as an educational resource with no clinical information or commercial affiliation.
Image Gallery

EARS
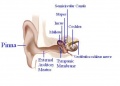
Anatomy of the ear
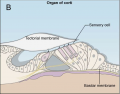
Histology of Inner Ear
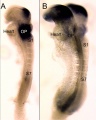
Pharyngeal arches one and two in mice embryo

The image depicts the development of the pinna through the fetal development stages
A view from inside the tympanic cavity of Lagenorhynchus obliquidens (Pacific White-sided Dolphin)
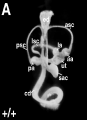
Wild-type inner ear showing normal morphology
Early expression of Pax2 and Pax8 compared
Otic placode of an embryo

Recent model related to sensory fate.
Zebrafish otic vesicle
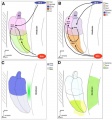
Representative expression patterns of genes controlling cochlear and vestibular specification.
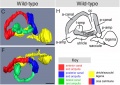
Wild-type semi-circular canal structure of zebrafish

Stereocilia bundles in the normal cochlea
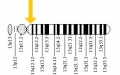
Mutation on GJB2 gene
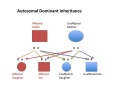
Dominant Inheritance
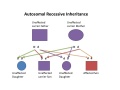
Autosomal Recessive inheritance
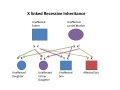
X-linked Recessive inheritance

Scanning electron microscopy, showing hair cells of the basal and middle cochlea
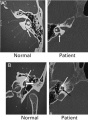
Pendred Syndrome

Goldenhar Syndrome
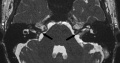
Bilateral Stenosis of Internal Auditory Canal
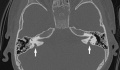
Enlarged Vestibular Aqueduct

Microtia
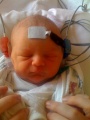
Testing the hearing of an infant - Auditory Brainstem Response test

Behind-the-ear (BTE), in-the-ear (ITE), in-the-canal (ITC), and completely-in-canal (CIC) hearing aids
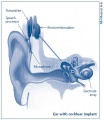
Cochlear Implant

Atoh1-cre is expressed in hair cells and causes transient limited expression of Atoh1 in CKO ears

Conditional deletion of Atoh1 results in death of organ of Corti cells and patchy Myo7a-positive presumptive hair cells which are innervated by many nerve fibers.

Aquaporins 1, 4, and 5 in the inner ear (A-C) and middle ear (D-G) – staining by AQP. H: KCNJ10 also stained the middle ear epithelium. Negative controls are displayed as inset pictures
--Mark Hill 12:22, 15 August 2012 (EST) Please leave the content listed below the line at the bottom of your project page.
2012 Projects: Vision | Somatosensory | Taste | Olfaction | Abnormal Vision | Hearing