2011 Group Project 3
| Note - This page is an undergraduate science embryology student group project 2011. |
Klinefelter's Syndrome
Introduction

Klinefelter's syndrome, first described in 1942 by Harry F. Klinefelter[1], is caused by the addition of one or more X chromosome(s) in affected males . He depicted a disorder characterised by gynecomastia and a very specific type of hypogonadism, as well as an absence of spermatogenesis.
It is still largely associated with these defects, the most common including reduced fertility and hypogonadism. A high proportion of affected men may remain asymptomatic their whole lives, this is because the severity of the disorder differs greatly from person to person[2].
The extra chromosome present is largely due to a genetic abnormality known as non-disjunction that can occur during either mitosis or meiosis, this is described in more detail below and portrayed in Figure 1. It has also been suggested to be the most common disorder associated with non-disjunction [2]
There are a number of diagnostic techniques currently used to determine accurate and early identification of the syndrome. This is particularly useful to encourage the implementation of the best treatment plan to manage the symptoms of the syndrome[3]. The phenotype of the syndrome differs significantly through the different stages of life, and a particular stage will correspond to the best management protocol for that point.
History

Klinefelter syndrome (KS) was first described by Harry F. Klinefelter and his colleagues in 1942. Their observations of nine patients where characterised by a number of peculiar symptoms; gynecomastia, azoospermia, hyalinised and small testes, absent spermatogenesis, elevated levels of follicle-stimulating hormone (FSH) and hypogonadism[4] [2]
In 1956, an investigation was carried out with 7 patients with KS that had the buccal smears that demonstrated Barr bodies . However, the cause of the syndrome remained unknown until 1959, when Jacobs and Strong discovered that a patient with KS had 47 chromosomes, including an extra X chromosome in the karotype of the patient [4]. As recorded in their article 'A case of human intersexuality having a possible XXY sex-determining mechanism';
| “…There are strong grounds, both observational and genetic, for believing that human beings with chromatin-positive nuclei are genetic females having two X chromosomes. The fact that this patient is chromatin-positive and has an additional chromosome within the same size range as the X, as well as an apparently normal Y, makes it seem likely that he has the genetic constitution XXY”[5] |
This discovery confirmed that the Barr bodies seen in patients with KS corresponds to an extra X chromosome[5]. In 1966, Harry F. Klinefelter reported that the extra X chromosome results from either meiotic nondisjunction or anaphase lag[6].
The ‘prototypic’ man with KS was initially described as tall, with narrow shoulders, broad hips, sparse body hair, gynecomastia, small testes, androgen deficiency and reduced verbal intelligence[4]. However, a few years after the syndrome was described Heller and Nelson reported that the gynacomastia was not a necessary part of the syndrome, even though it occurred in about 75% of the patients which they observed. The hallmarks of the syndrome were then thought small testes, sterility and increased excretion of follicle stimulating hormone[6]. Extensive studies of these patients during their adolescence illustrated the various personality traits, which can be handled by proper counselling. Similar to today, Harry Klinefelter reported that most patients with this condition were not diagnosed until early adult life, when counselling may be less rewarding.
Below is a timeline highlighting all the major advancements and developments in KS.
Timeline
| Year | Discovery |
| 1942 | Dr. Harry Klinefelter and his co-workers at Massachusetts General Hospital in Boston published a report on 9 men describing the signs and symptoms of KS. The ‘prototypic’ man with KS was described as tall, with narrow shoulders, broad hips, sparse body hair, gynecomastia, small testes, androgen deficiency and reduced intelligence. [4] |
| 1949 | Barr and Bertram noticed positive chromatin material in KS patients.It was a dense chromatin mass which they later termed, Barr body. |
| 1956 | The discovery of Barr bodies as a result of the new development of Buccal smears |
| 1959 | Jacobs and Strong discovered that it was a chromosomal disorder, with the appearance of an extra X chromosome. |
| 1960s | Development of chromosome banding techniques. [7] |
| 1962 | Maclean and Mitchell’s studies on inmates in prisons and institutions for mental health revealed an increased risk of psychiatric disorders, mental retardations and criminal behaviour in patients with KS. [4] |
| 1966 | Dr. Harry Klinefelter reports that the extra X chromosome results from either meiotic non-disjunction or anaphase lag. [6] |
| 1970 | Rozen et al reported that approximately 1% of all individuals institutionalised with mental retardation have an XXY karotype. [8] |
| 1970s | A number of centres began screening newborns for sex chromatin abnormalities. |
| 1986 | Dr. Harry Klinefelter described that the hallmarks of the syndrome are now small testes, sterility and increased excretion of follicle stimulating hormone. [6]
He also reported that most patients with this condition were not diagnosed until early adult life, when counselling may be less rewarding. [6] Treated hypogonadism with injected testosterone, and thought this also aided personality abnormalities in adolescent patients. [6] |
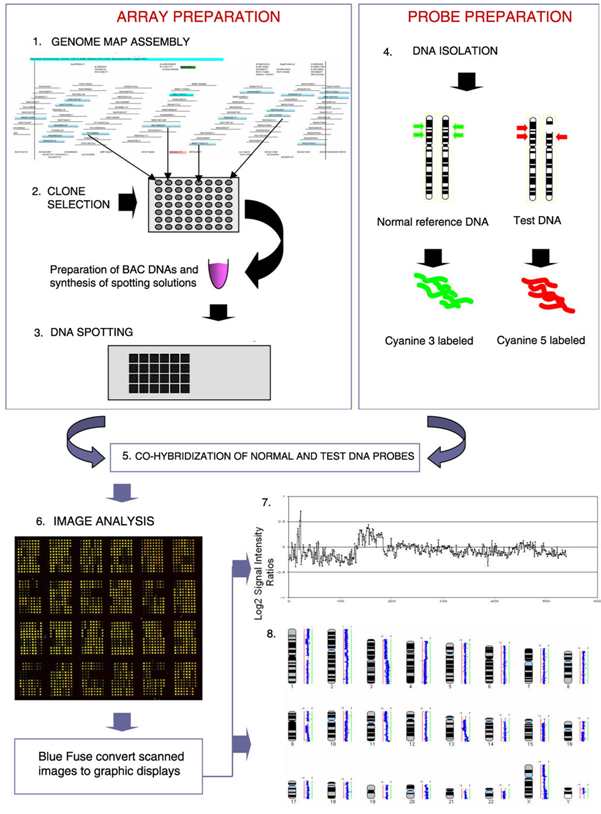
| 1992 | The Comparative Genomic Hybridization (CGH) analysis was developed as a genome wide screening strategy for detecting DNA copy number imbalances.[9] A classic array-CGH experiment is shown in Figure 2. |
| 1995 | Reiss et al., found that half all mental retardation in males originates from a defective gene on the X chromosome. Thus, the X chromosome comprises the genes involved in human cognition. [10] |
| 1998 | Smyth and Bremmer concluded that XXY karotypes occurs in 1 in 500 live male births and is the most common type of human chromosome anomaly. [11]
They also hypothesised that the characteristics features of KS originate from genes escape inactivation and are expressed in excess. |
| 2002 | Crow et al., suggested that at least one gene or genes in the X-Y homologous regions of the sex chromosomes which escape’s normal X- inactivation are crucial for language functioning.[12] |
| 2010 | Stefano Gambardella et al., designed a targeted array called GOLD (gain or loss detection) Chip which detects unexpected major chromosome imbalances. [13] |
Epidemiology
One of the most common disorders of sex chromosomes in humans is Klinefelter’s syndrome (KS), otherwise known as 47,XXY gene mutations. This is prevalent in around 1 in 500 males[14]. There can also be variations of this genetic condition, and these variations are referred to as chromosomal aneuploidies. The chromosomal variations are present within 1 in 50 000 male births, so are much rarer than 47,XXY mutations. It is said that males born with KS often go through life without being karyotyped, meaning that they are left undiagnosed[15]. In around 80% of cases, the karyotype for KS is shown in every cell of the body. The age of the mother and father at the time of conceiving a child has no relation at all to whether a child will be born with the condition.
A link was found between increased risk of mortality and KS. There was “a significant increase in mortality risk of 40% (hazard ratio, 1.40; 95% confidence interval, 1.13–1.74), corresponding to a significantly reduced median survival of 2.1 yrs.’’ [16]The increased mortality was because of infections, neurological, circulatory, pulmonary, and urinary tract diseases which people with KS are more susceptible too. There are studies currently being conducted into whether socioeconomic background increases the risk of a child developing KS[16].
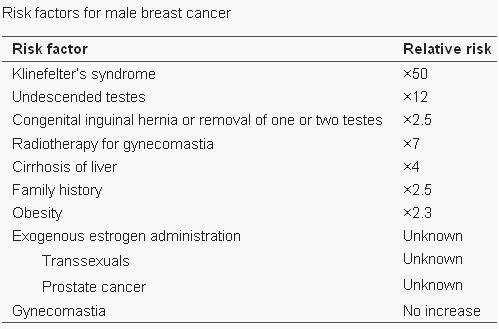
Seizures can typically occur, and when seizures occur in males with KS, it usually happens between 3 months and 3 years of age. Neuro-imaging tests have failed to identify the cause of the seizures[17]. It is very difficult to diagnose a child with KS immediately, since many of the symptoms that are exhibited in childhood may be due to other factors, such as shyness, stress, and social phobia. Figure 3 is a graph adapted from recent studies, demonstrating the emotional response to stimuli of men with KS compared to normal males. It has also been suggested that men with KS are 50% more at risk of being diagnosed with breast cancer, as shown in Figure 4.[18]
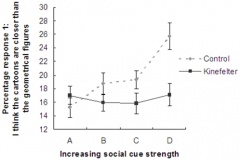
Figure 3: The emotional response and comprehension of men with Klinefelter’s Syndrome differs from that of normal men
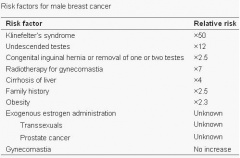
Figure 4: Risk factors associated with male breast cancer


Aetiology
Chromosome abnormalities have a high incidence in humans. The most common type is aneuploidy, which is the loss (monosomy) or gain (trisomy) of an entire chromosome [19]. Aneuploidy’s occur in approximately 5% of pregnancies which survive long enough to be seen and approximately 10-25% of all fertilised human oocytes are either monosomic or trisomic [19]. Trisomies are incapable of normal development, the consequences are less severe for the sex chromosomes than the autosomes, leading to enhanced sex chromosomes among live-borns in comparison with autosomal trisomies. Abnormal chromosome distribution, is the result of one of two mechanisms; non-disjunction and anaphase lagging. However, Klinefelter’s syndrome(KS) is more commonly caused by non-disjunction.[20]
Non-Disjunction

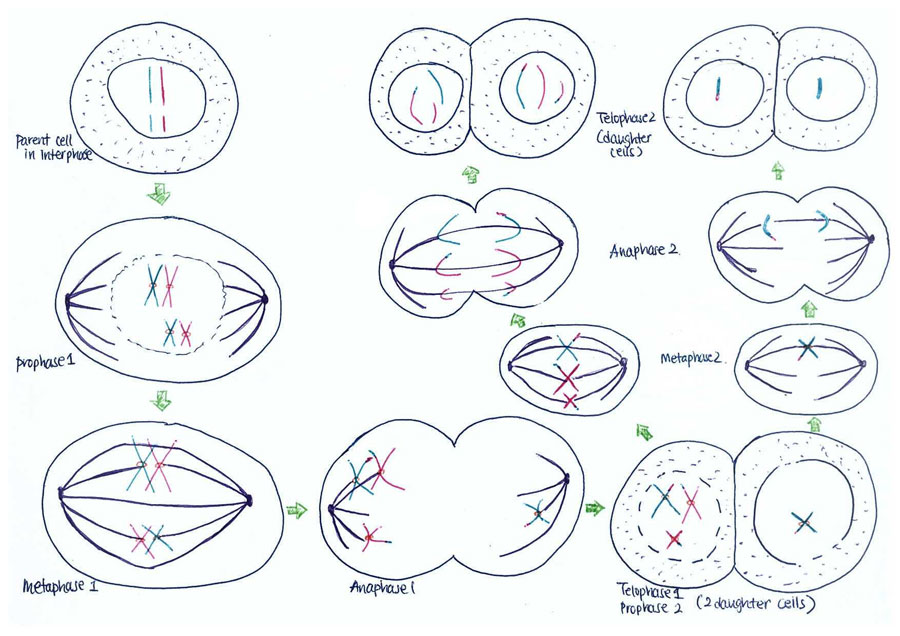
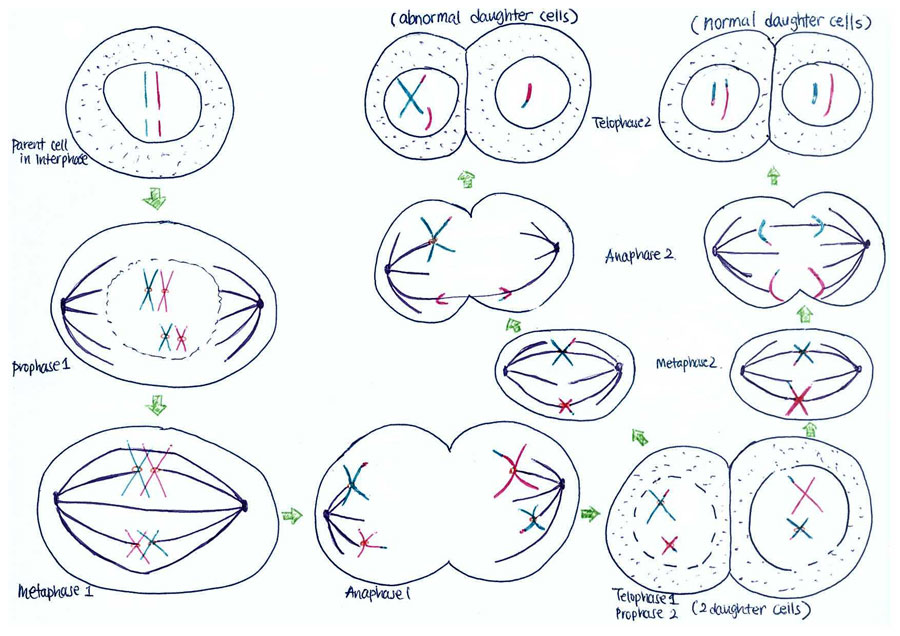
Non-disjunction is the failure of chromosome pairs to separate during the first and second meiotic divisions, as shown in Figure 5. Maternal XXY can be caused by non-disjunction during the first and second meiotic divisions, however, XXY of paternal origin can only occur during the first meiotic division. In the great majority of trisomies the additional chromosome is of maternal origin and results from an error during the first meiotic division as seen in Figure 5. However, the 47,XXY has been extensively studied and there is a 50-60% chance of it being from a paternal origin and 40-50% from a maternal origin[21]. The nature of the errors by which maternal and paternal XXYs can arise are extremely diverse. Studies have established that increased maternal age has adverse affects on pregnancy and an increased chance of anueploidy.[22] The process of meiosis serves to generate haploid gametes through a specialised cell division process, consisting of two stages of cell division, MI and MII. During prophase of MI the pairs of homologous chromosomes synapse and undergo recombination, with chiasmata being formed at the sites of exchange. Their purpose is to link together the homologues and therefore play a crucial role in the proper disjunction of chromosomes in the first meiotic division [19].
This more common aberrant cell division takes place during gametogenesis. More specifically, it is the event of the homologous chromosome or sister chromatid failing to separate normally.[4] So like anaphase lagging, the distribution of genetic content in the daughter cells is uneven. In addition, it can also occur in both of the meiotic divisions, leading to variants of KS with a greater number of X chromosomes. Some examples of these variants are 48,XXXY and 48,XXXXY. Furthermore, non-disjunction can also take place in postzygotic division. [23]
As depicted in Figure 7, when non-disjunction takes place in the first meiotic division, a homologous pair of chromosomes is not separated in the anaphase 1. The result is one daughter cell having three chromosomes while the other has only one by the end of meiosis -1. The daughter cell with one chromosome is not viable. When the daughter cell with three chromosomes is followed through meiosis two, its own progeny can be seen to include either a parental and maternal X chromosome or two copies of the maternal X chromosome.
However, if non-disjunction occurs in the second meiotic division, then the abnormal separation of genetic material takes place in anaphase 2. This is seen in Figure 8. By the end of the second meiosis one daughter cell contains both sister chromatids of the maternal X chromosome, while the other daughter cell does not contain that maternal genetic material.
The consequence of non-disjunction is that an XX ovum or a XY sperm will be produced. This gamete will then go on to be fertilised by a normal Y sperm or an X ovum respectively and conclusively produce a XXY zygote, which is the hallmark of KS.
Animation Meiotic Non-disjunction - Meiosis I
Animation Meiotic Non-disjunction - Meiosis II
- Images of Non-disjunction
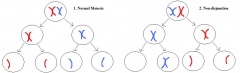
Figure 6: Meiotic Non-disjunction in Comparison with Normal Disjunction.
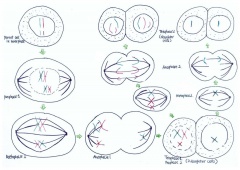
Figure 7: Nondisjunction of Homologous Chromosomes in Meiosis 1.
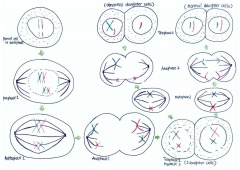
Figure 8: Nondisjunction of Homologous Chromosomes in Meiosis 2.



Anaphase Lagging
In the anaphase phase of cell division, spindle fibers attach to sister chromatids and separate them. However, in the abnormal event of anaphase lagging, some spindle fibers are missing. Because of this, sister chromatid will not be successfully separated but together will be incorporated into a single daughter nucleus. As a result, the daughter cell will have a supernumerary chromosome.
Pathophysiology
Although extensive studies have been made to discover the pathophysiology of Klinefelter’s syndrome(KS), i.e. the link between the supernumerary X and the phenotype, it remains largely unclear. Postulations of the pathophysiology of KS have been derived from comparing the phenotype 47,XXY with other sex chromosome aneuploidies. The correlation with this phenotype is established with higher order sex chromosome aneuploidies , such as 48,XXXY. However several genetic mechanisms may explain the variability of the phenotype, clinical features, life circumstances, life expectancy and fertility [24]. Meiotic failure has essential roles in the parental origin of the X chromosome, gene-dosage effects in conjunction with (possibly skewed) X chromosome inactivation (XCI) and especially spermatogenesis[24]. Typically, when two or more X chromosomes are present in a cell, as in healthy females or sex chromosome disorders like 47,XXY, only one is active. The additional X chromosome is mostly inactive and the X chromatin is perceived as a Barr body in the periphery of the cell nucleus[25].
In addition, both X and Y chromosomes carry short regions of homology termed pseudoautosomal regions (PAR) [26] which remain active in men and woman [27]. They behave as an autosome and function to allow X and Y chromosomes to pair and properly segregate during meiosis in males. Some genes in the X chromosome which are not homologous to the Y chromosome can escape inactivation and are functionally duplicated in KS males[27]. PAR1 comprises 2.6Mb of the short arm tips of both X and Y chromosomes, PAR2 at the tips of the long arms spans a much shorter region of 320kb. Since the sequencing of the X chromosome, it has been noted that PAR1 contains at least 24 genes whereas in PAR2 only 4 have been identified and that 10% of X chromosomal genes are specifically expressed in the testis[24].
XCI can occur randomly or by imprinting, this is where the paternal X chromosome is silenced in the preimplantation embryo and extraembryonic tissue. Random XCI occurs in the epiblast and can inactivate either the maternally or paternally inherited X chromosome, resulting is an active and inactive chromosome, which is transmitted to descendant cells. Some studies have analysed the random XCI in KS patients, and found that there is a skewed inactivation of one allele and this has been detected in a variety of cases. Many other studies have been conducted and this has led to skewed XCI ranges from around 10-40% of cases in KS patients[24].
The X-inactivation centre which initiates XCI contains the X (inactive) specific transcript (XIST). XIST encodes an untranslated RNA which can coat and silence the X chromosome. In addition to non-coding transcripts, XCI involves chromatin modifiers and factors of nuclear organisation. Together these lead to a changed chromatin structure and spatial reorganisation of the silenced X chromosome[24]. However, PARs are not inactivated to achieve the same gene-dosage in both sexes. PARs are important because they allow the correct order and fusion of X and Y chromosomes. Clearly, in the case of KS, this fusion is not correct since XXY results and there is the inclusion of an extra X chromosome in the genotype[28]. Therefore, the pseudoautosomal region has a mutation. This mutation can lead to other various genetic disorders as well as KS and the mutation being present in PAR1 or PAR2 can have a similar effect. These mutations will proceed to give rise to the clinical manifestations of KS [28].
The cardinal features of KS will arise from the error in germ cell survival in the aneuploid testis in the embryo. When the child reaches puberty, the germ cells are destroyed instead of proliferating the seminiferous tubules. This causes some of the symptoms of KS, such as azoospermia with the formation of small testis. For more information, see the table on signs and symptoms .

Signs and Symptoms
Symptoms may differ slightly depending on the stage of development. Screening is recommended when a combination of the following signs of Klinefelter’s syndrome(KS) are observed.
| Age | Signs and Symptoms | Image and Links | |
| Birth |
|
||
| Infancy |
| ||
| Childhood |
| ||
| Puberty |
|
||
| Adulthood |
|




Link to an article which has images which compares testicular specimens from various age groups
Diagnosis
Karyotype
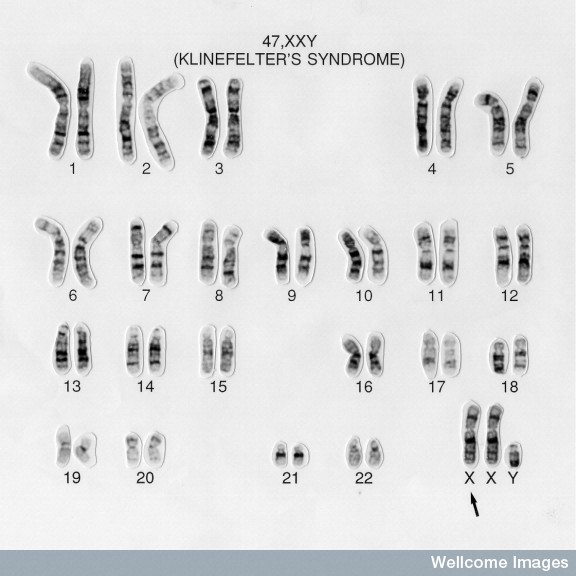
Karyotyping is a process where the number of chromosomes and their structures are observed for any abnormalities. In a healthy male subject, the karyotype analysis will show 46,XY. But in a Klinefelter's syndrome(KS) patient, the karyotype is often observed to be 47,XXY. The featured image is an example of a typical karyotype of a KS patient.
Movie on the process of karyotyping
Statistics
The chances of diagnosing KS are, unfortunately, very low. In one study, only 36% of KS patients are diagnosed, of which 10% of the KS patients being diagnosed prenatally and 26% post natally. This shows that diagnosis methods are not easily accessed and performed. [36]
Prenatal Diagnosis methods
Prenatal diagnosis is most common achieved by two methods, either by amniocentesis or chorionic villus sampling (CVS). As both are invasive procedures, they have considerable risks. Most often these diagnosis methods are performed because other genetic defects are suspected, such as Down ’s syndrome, because of an advanced maternal age. Therefore, the diagnosis of KS is often considered a chance finding.
Amniocentesis can be done from approximately 14 weeks gestation, it involves the sampling of amniotic fluid. The amniotic fluid contains fetal DNA and cells, proving very useful for such testing.

A needle is passed into the mother’s abdomen and into the amniotic cavity. This process is always used in conjunction with an ultrasound, to ensure the best conditions are available, ie fetal position in womb. [37]
For CVS, a catheter is passed through the mother’s vagina and into the uterus using ultrasound imaging. From here, cells from the chorionic placenta can be sampled for future testing. This can be carried out in the first trimester of pregnancy, a significant benefit over amniocentesis. However, there is a risk of miscarriage with this test, especially when compared to the process amniocentesis. [38]
The cells from both samples are then cultured until there are sufficient numbers of cells. An enzyme is then added that halts proliferation at a point when the chromosomes are condensed. These chromosomes are then stained and, using a light microscope, any abnormalities can be identified. [39]
Diagnosis at birth
Although this rarely takes place, some clinical features may make physicians suspect and ask for a karyotype analysis. Some abnormalities include:
• genital abnormalities (eg. cryptorchidism, small penis, scrotum bifidus and hypospadias)
• fifth finger clinodactyly
• cleft palate
• inguinal hernia [40]
Postnatal Diagnosis Methods
_assay.JPG)
Postnatal diagnositic test for KS are often performed because physicians may suspect it because of clinical findings. As described in the ‘Signs and Symptoms’ section, some clinical findings may include patients suffering from infertility, learning disabilities and osteoporosis. Furthermore, a blood test of a KS patient may show high levels of follicle stimulating hormone, luteinizing hormone and low levels of testosterone. [41]
Postnatal karyotyping is a process where a blood sample is analysed. Leukocytes from the patient’s blood sample are separated and then cultured. They are halted in the cell cycle when the chromosomes are condensed and can be stained. A photograph is taken of the chromosomes from one cell. Then they are organized according to size, number and structure and then observed for abnormalities.[42][43]
Management
Androgen Therapy
Androgen therapy involves the replacement of testosterone. This is primarily given to stimulate the onset of puberty in affected males. Ideally, testosterone is given from the age that puberty usually occurs, in order to encourage normal development. In addition to this, it assists in treating or preventing some of the more typical clinical presentations of this disorder. Testosterone replacement encourages secondary sexual attributes, and helps ensure standard bone and muscle mass[44].
Samango-Sprouse et al found that the initiation of androgen therapy early in life was highly associated with improvments in speech and cognition, and other neurogical development. This research was based around children with severe KS - 49,XXXXY. This gives much hope to the prospect of early treatment resulting in normal development of those afflicted with KS. [45]

However, it has also been associated with a decrease in fertility, especially if given early in life. Premature treatment has been suggested to result in delayed puberty and abnormal physical development during this period[46]. It is also recommended to stop testosterone replacement a few months prior to the administration of infertility treatment[46]. [47]
Fertility
Aromatase inhibitors can be administered to men, in order to lower intratesticular estradiol levels. This is thought to encourage the production of testosterone and activate spermatogenesis[48]. There are two main methods used to treat non-obstructive azoospermia, microdissection testicular sperm extraction (TESE) and conventional TESE. These are methods of extracting what sperm is present in the testes for use in in vitro fertilisation (IVF). It has been shown that microdissection TESE has a higher rate of extraction, and allows for minial testicular damage[49], and so conventional TESE is slowly being replaced by microdissection TESE. The most common IVF technique used in these situations is intracytoplasmic sperm injection, where a single sperm is injected into a single oocyte. This means that for the highest chance of success, the extraction of both the sperm and the egg need to be well timed[50]. Before surgery, men are usually administered aromatase inhibitors for a few months. This is to restore the ratio of testoserone to esradiol back to normal levels, and to encourage the amount of viable sperm present[48].
Other Similar Defects
| Abnormality | Description | Similarities | Differences | Image |
| Turner Syndrome (XO) | This is a condition where a female only has one sex chromosome (45,XO). Girls with Turner syndrome lack certain characteristics, such as full grown ovaries, metabolic problems, short stature and decreased life expectancy [51].
In embryology, the embryo may present with a web-like neck, low birth length and lymphedema of the dorsum of hands and feet. There may also be a delay in puberty because of gonadal dysgenesis. As well as all this, there may also be congenital defects of the heart, kidney and autoimmune system [52]. |
|
|
|
| 47,XYY | This is a condition where a male inherits an extra Y chromosome. The additional Y chromosome is from the father (paternal origin) and there is existing evidence that spermatocytes with additional Y chromosomes are selected against during gametogenesis [54]. This condition only affects males.
This condition is not usually passed on from parents to offspring, and it has been shown that the sperm of 47,XYY males has the normal karyotype [55]. |
|
|
|
| 48,XXYY | 48,XXYY, frequently referred to as another variant of KS, is an anomaly whereby males have an extra X and Y chromosome. Sometimes, renal clearance can be affected by this condition. This is caused by low levels of serum urate in conjunction with high levels of renal urate clearance [57]. |
|
|



Current Research
Neural systems for social cognition in Klinefelter syndrome(KS) (47,XXY): evidence from fMRI; 2011

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
It is suggested that the addition of an X chromosome has a significant impact on neural systems. The X chromosome contains a large number of genes responsible for neural system development, and so KS was the ideal model for the observation of neural development abnormalities. van Rijn et al used fMRI (functional magnetic resonance imaging) to observe the activity in specfic parts of the brain. People with KS are generally socially disadvantaged, and so a link was investigated between neural activity and social cues.
It was found that KS can be associated with decreased activity in specific neural systems. This also advocates the possibility of using KS as a model for further study of the X chromosome. [60]
Insights into the pathogenesis of XXY phenotype from comparison of the clinical syndrome with an experimental XXY mouse model; 2010
Even after all we now know about KS, a lot still remains a mystery. This is particularly due to the absence of a commonly used animal model. Lue et al investigated the ultimate similarities of a mouse 41,XXY model (as opposed to the human 47,XXY), and it's use in further research. It is important to have an animal model that largely resembles the human syndrome or disease. Lue et al used this model to compare phenotypic similaries between the two, in regards to hormonal imbalances and effect of additional X chromosome genes.
By the comparison of the animal model and human representative, they found that clinical symptoms and phenotype typical in KS are likely due to an avoidance of the inactivation of the X chromosome. [61]
Expression of selected genes escaping from X inactivation in the 41, XXY* mouse model for Klinefelter’s syndrome(KS); 2010
It still remains a debate as to the exact molecular and genetic actions that result in the presentation of KS. It has been suggested that some of the genes on the X chromosome escape inactivation and so result in the clinical symptoms of KS. Werler et al explored this theory using two mouse models (41,XXY and 41,XXY*) and epigenetics. Out of the genes that were obseved in this study, they all seemed to undergo escape from X chromosome inactivation similarly to female mice.
There were also noticable tissue differences, wherein gene expression in the brain was significantly different to that of the liver and kidney. In the brain there was substantial upregulation of these "X-linked excapee genes". This is suggested to directly affect the phenotypic presentation of 47,XXY syndrome. [62]
Related Links
Internal
- Oocyte Development | This page describes the normal development of an oocyte, also discussing the abnormalities associated such as non-disjunction.
- Gametogenesis | This page, similarly, describes the normal development of the gametes and the associated abnormalities. The relate to the development of Klinefelter's syndrome.
- Cell Division - Meiosis | This page describes the process of meiosis and the possible associated irregularities such as non-disjunction.
- Abnormal Development - Genetic | This page largely discusses the possible genetic abnormalities and their formation such as aneuploidy, which is the cause of Klinefelter's syndrome.
- Genital Development | Irregular genital development is one of the major symptoms associated with Klinefelter's syndrome. This page gives an overview of normal genital development and the associated abnormalities.
External
- Andrology Australia | This page gives a concise summary of Klinefelter's syndrome.
- Information booklet | This is a detailed information booklet regarding Klinefelter's syndrome, published by the Australian Paediatric Endocrine Group
Glossary
- Acromegaly - This is a condition caused by abnormal hormone production from the pituitary gland, resulting in altered growth of hands, feet, and face
- Anaphase lag - Describes a chromosome which is not incorporated into the new cell in the second stage of mitosis(anaphase) due to it ‘lagging’, resulting in gametes lacking a sex chromosome
- Androgen - A male sex hormone, ie testosterone
- Aneuploidy - Abnormal number of chromosomes
- Azoospermia - Occurs when males have little to no motile sperm in the semen
- Barr bodies - Inactivated X chromosome in females due to sex being determined by W or Y chromosomes instead of XY
- Buccal mucosal cells - Cells of the oral cavity that secrete mucus
- Chromatin - The genetic material that forms chromosomes, it is also composed of DNA
- Cryptorchidism - 1 or 2 testes have not dropped into the scrotum from the abdominal cavity
- Dysgenesis - Defective or abnormal development of an organ, especially of the gonads
- Gametogenesis - Biological process by which haploid or diploid cells undergo division and/or differentiation to form mature haploid gametes.
- Gonadotropin - Protein hormones secreted by gonadotrope cells of the pituitary gland.
- Gynecomastia - Is the abnormal development of large mammary glands in males which give large breasts.
- Hyalinised - The state of something being hyaline (clear and translucent)
- Hypogonadism - Occurs when the sex glands produce little to no hormones.
- Hypotonia - reduced muscle tone
- In vitro fertilisation - The process of fertilising an oocyte by sperm outside the womb.
- Karyotype - Number and/or appearances of chromosomes in a eukaryotic cell nucleus
- Leukocytes - White blood cells which are cells of the immune system
- Libido - sexual desire
- Meiosis - The process of cell division of germ (sex) cells resulting in 4 daughter cells.
- Mosaicism - The presence of two or more genetically different cells in one organism
- Non-disjunction - The failure of chromosomes to separate properly during cell division. This results in abnormal chromosome number in daughter cells.
- Pituitary gland - Endocrine gland that secretes hormones that regulate homeostasis.
- Post zygotic non-disjunction - This occurs when chromosomes do not separate during mitosis, and the cells are not removed by the usual 'proof-reading' mechanisms.
- Prepulse - A weak stimulus that inhibits a reaction to the following stimulus.
- Seminiferous tubule - Long, thread like tubes found in the testes and are the specific location of meiosis in the body.
- Sertoli cells - Cell of the testes that is part of the seminiferous tubule. It is activated by Follicle Stimulating Hormone
- Smooth pursuit eye movements - A slow and trailing eye movement which allows the moving object of attention to remain in line with the fovea.Definition from the Neuroscience textbook
- Spermatogenesis - Process by which male germ cells undergo division and produce a number of cells referred to as spermatogonia, through which spermatocytes are derived from.
References
- ↑ Klinefelter HF, Reifenstein EC & Albright F. Syndrome characterized by gynecomastia, aspermatogenesis without a-Leydigism, and increased excretion of follicle-stimulating hormone. American Journal of Clinical Dermatology 1942; 2: 615–627.
- ↑ 2.0 2.1 2.2 Wikström AM, Dunkel L. Klinefelter syndrome. Best Pract Res Clin Endocrinol Metab: 2011, 25(2):239-50
- ↑ <pubmed>20392711</pubmed>
- ↑ 4.0 4.1 4.2 4.3 4.4 4.5 <pubmed>17415352</pubmed>
- ↑ 5.0 5.1 <pubmed>13632697</pubmed>
- ↑ 6.0 6.1 6.2 6.3 6.4 6.5 <pubmed>3529433</pubmed>
- ↑ <pubmed>5640698</pubmed>
- ↑ <pubmed>4398603</pubmed>
- ↑ <pubmed> 1359641</pubmed>
- ↑ <pubmed>7585014</pubmed>
- ↑ <pubmed>9645824</pubmed>
- ↑ <pubmed> 15729733</pubmed>
- ↑ Stefano G., Erika C., Francesca M., Giusy S., Francesca G., Michela B., Anna M. N., Antonio N., Ercole B., Laura B. and Giuseppe N.(2010) Design, Construction and Validation of Targeted BAC Array-Based CGH Test for Detecting the Most Commons Chromosomal Abnormalities. ‘’Genomic Insights.’’ 3:9-21
- ↑ <pubmed>17062147</pubmed>
- ↑ <pubmed>9160389</pubmed>
- ↑ 16.0 16.1 <pubmed>15292313</pubmed>
- ↑ <pubmed>20438626</pubmed>
- ↑ <pubmed>20463819</pubmed>
- ↑ 19.0 19.1 19.2 <pubmed>12926525</pubmed>
- ↑ Cynthia M. Smyth, William J. B. Klinefelter Syndrome Arch Intern Med. 1998;158:1309-1314
- ↑ <pubmed>11426451</pubmed>
- ↑ <pubmed>11582569</pubmed>
- ↑ Bandmann, H. J., Breit, R., & Perwein, E., (1984). Genetics and Cytogenetics of Klinefelter's Syndrome: Karyotype of Klinefelter's Syndrome and Its Variants. In H. J. Bandmann, B. Reinhardt, & E. Perwein, Klinefelter's Syndrome (1, pp. 1-229). New York, America: Springer-Verlag.
- ↑ 24.0 24.1 24.2 24.3 24.4 <pubmed>20228051</pubmed>
- ↑ <pubmed>21521364</pubmed>
- ↑ <pubmed> 20228051</pubmed>
- ↑ 27.0 27.1 <pubmed>21521364</pubmed>
- ↑ 28.0 28.1 <pubmed>16601196</pubmed>
- ↑ http://www.genome.gov/19519068
- ↑ http://emedicine.medscape.com/article/945649-clinical#a0217
- ↑ Schlatt S, Hillier SG &Foresta C, Klinefelter’s syndrome: from chromosome to clinic Molecular Human Reproduction 2010;16: 373– 374
- ↑ <pubmed>19221583</pubmed>
- ↑ Aksglæde L, Skakkebæk NE, Almstrup K, Juul A. Clinical and biological parameters in 166 boys, adolescents and adults with nonmosaic Klinefelter syndrome: a Copenhagen experience.Acta Pædiatrica 2011, 100(6);793–806
- ↑ Daniel JW, MAJ, MC, USAF, and Maximilian M, M.D. Klinefelter Syndrome Am Fam Physician. 2005 Dec 1;72(11):2259-2262.
- ↑ 35.0 35.1 35.2 <pubmed>21655260</pubmed>
- ↑ Abramsky L, Chapple J. 47,XXY (Klinefelter syndrome) and 47,XYY: estimated rates of and indication for postnatal diagnosis with implications for prenatal counselling. Prenat Diagn 1997;17:363 – 368
- ↑ <pubmed>15758614</pubmed>
- ↑ <pubmed>21413037</pubmed>
- ↑ <pubmed>135003</pubmed>
- ↑ Lee YS, Wai Fun Cheng A, Ahmed SF, Shaw JN, Hughes AI. Genital anomalies in Klinefelter’s Syndrome. Horm Res 2007;68:150 – 155.
- ↑ http://tidsskriftet.no/article/1695231 Week. 11 - 29 May 2008 Journal of Medical Unite 2008: 128:1281-3
- ↑ <pubmed>15183749</pubmed>
- ↑ http://www.genome.gov/19519068
- ↑ <pubmed>20482304</pubmed>
- ↑ <pubmed>21362043</pubmed>
- ↑ 46.0 46.1 <pubmed>18832949</pubmed>
- ↑ <pubmed>21655260</pubmed>
- ↑ 48.0 48.1 <pubmed>11792932</pubmed>
- ↑ <pubmed>21811543</pubmed>
- ↑ <pubmed>21716935</pubmed>
- ↑ <pubmed>21454226</pubmed>
- ↑ <pubmed>21271130</pubmed>
- ↑ <pubmed>21818630</pubmed>
- ↑ <pubmed>10545600</pubmed>
- ↑ <pubmed>18037669</pubmed>
- ↑ <pubmed>20014371</pubmed>
- ↑ <pubmed>3714610</pubmed>
- ↑ <pubmed>18481271</pubmed>
- ↑ <pubmed>8389624</pubmed>
- ↑ <pubmed>21737434</pubmed>
- ↑ <pubmed>21217605</pubmed>
- ↑ <pubmed>21241365</pubmed>
2011 Projects: Turner Syndrome | DiGeorge Syndrome | Klinefelter's Syndrome | Huntington's Disease | Fragile X Syndrome | Tetralogy of Fallot | Angelman Syndrome | Friedreich's Ataxia | Williams-Beuren Syndrome | Duchenne Muscular Dystrolphy | Cleft Palate and Lip