The printable version is no longer supported and may have rendering errors. Please update your browser bookmarks and please use the default browser print function instead.
Introduction
|

|
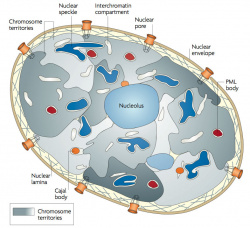
| Chromosome territories (interphase)
|
Chromosome (Chromatin) structure (mitosis)
|
Some Recent Findings
- Monosomy 7 in Pediatric Myelodysplastic Syndromes[1] "Myelodysplastic syndromes (MDS) in children and adolescents are a rare heterogeneous group of clonal stem cell disorders. Complete or partial loss of chromosome 7 constitutes the most common cytogenetic abnormality encountered in any type of childhood MDS, is associated with more advanced disease, and usually requires a timely allogeneic stem cell transplantation. This article provides insights into the current understanding of the genotype, phenotype, and clonal evolution patterns in pediatric MDS associated with loss of chromosome 7."
- The enigma of monosomy 7[2] "Since a report of some 50 years ago describing refractory anemia associated with group C monosomy, monosomy 7 (-7) and interstitial deletions of chromosome 7 (del(7q)) have been established as one of the most frequent chromosomal aberrations found in essentially all types of myeloid tumors regardless of patient age and disease etiology. In the last century, researchers sought recessive myeloid tumor-suppressor genes by attempting to determine commonly deleted regions (CDRs) in del(7q) patients. However, these efforts were not successful. Today, tumor suppressors located in 7q are believed to act in a haploinsufficient fashion, and powerful new technologies such as microarray comparative genomic hybridization and high-throughput sequencing allow comprehensive searches throughout the genes encoded on 7q. Among those proposed as promising candidates, 4 have been validated by gene targeting in mouse models. SAMD9 (sterile α motif domain 9) and SAMD9L (SAMD9-like) encode related endosomal proteins, mutations of which cause hereditary diseases with strong propensity to infantile myelodysplastic syndrome (MDS) harboring monosomy 7. Because MDS develops in SAMD9L-deficient mice over their lifetime, SAMD9/SAMD9L are likely responsible for sporadic MDS with -7/del(7q) as the sole anomaly. EZH2 (enhancer of zeste homolog 2) and MLL3 (mixed lineage leukemia 3) encode histone-modifying enzymes; loss-of-function mutations of these are detected in some myeloid tumors at high frequencies. In contrast to SAMD9/SAMD9L, loss of EZH2 or MLL3 likely contributes to myeloid tumorigenesis in cooperation with additional specific gene alterations such as of TET2 or genes involved in the p53/Ras pathway, respectively. Distinctive roles with different significance of the loss of multiple responsible genes render the complex nature of myeloid tumors carrying -7/del(7q)."
- Maintenance of Mest imprinted methylation in blastocyst-stage mouse embryos is less stable than other imprinted loci following superovulation or embryo culture[3] "Assisted reproductive technologies are fertility treatments used by subfertile couples to conceive their biological child. Although generally considered safe, these pregnancies have been linked to genomic imprinting disorders, including Beckwith-Wiedemann and Silver-Russell Syndromes. Silver-Russell Syndrome is a growth disorder characterized by pre- and post-natal growth retardation. The Mest imprinted domain is one candidate region on chromosome 7 implicated in Silver-Russell Syndrome. We have previously shown that maintenance of imprinted methylation was disrupted by superovulation or embryo culture during pre-implantation mouse development. For superovulation, this disruption did not originate in oogenesis as a methylation acquisition defect. However, in comparison to other genes, Mest exhibits late methylation acquisition kinetics, possibly making Mest more vulnerable to perturbation by environmental insult. In this study, we present a comprehensive evaluation of the effects of superovulation and in vitro culture on genomic imprinting at the Mest gene. Superovulation resulted in disruption of imprinted methylation at the maternal Mest allele in blastocysts with an equal frequency of embryos having methylation errors following low or high hormone treatment. This disruption was not due to a failure of imprinted methylation acquisition at Mest in oocytes. For cultured embryos, both the Fast and Slow culture groups experienced a significant loss of maternal Mest methylation compared to in vivo-derived controls. This loss of methylation was independent of development rates in culture. These results indicate that Mest is more susceptible to imprinted methylation maintenance errors compared to other imprinted genes." Epigenetics
|
| More recent papers
|
|
This table allows an automated computer search of the external PubMed database using the listed "Search term" text link.
- This search now requires a manual link as the original PubMed extension has been disabled.
- The displayed list of references do not reflect any editorial selection of material based on content or relevance.
- References also appear on this list based upon the date of the actual page viewing.
References listed on the rest of the content page and the associated discussion page (listed under the publication year sub-headings) do include some editorial selection based upon both relevance and availability.
More? References | Discussion Page | Journal Searches | 2019 References | 2020 References
Search term: Chromosome 7
|
| Older papers
|
| These papers originally appeared in the Some Recent Findings table, but as that list grew in length have now been shuffled down to this collapsible table.
See also the Discussion Page for other references listed by year and References on this current page.
|
Development Genes
WNT
| Human WNT Family
|
| Table - Human Wnt Family
|
Approved
Symbol
|
Approved Name
|
Previous
Symbols
|
Synonyms
|
Chromosome
|
| WNT1 |
Wnt family member 1 |
INT1 |
|
12q13.12
|
| WNT2 |
Wnt family member 2 |
INT1L1 |
IRP |
7q31.2
|
| WNT2B |
Wnt family member 2B |
WNT13 |
XWNT2 |
1p13.2
|
| WNT3 |
Wnt family member 3 |
INT4 |
"MGC131950, MGC138321, MGC138323" |
17q21.31-q21.32
|
| WNT3A |
Wnt family member 3A |
|
|
1q42.13
|
| WNT4 |
Wnt family member 4 |
|
WNT-4 |
1p36.12
|
| WNT5A |
Wnt family member 5A |
|
hWNT5A |
3p14.3
|
| WNT5B |
Wnt family member 5B |
|
|
12p13.33
|
| WNT6 |
Wnt family member 6 |
|
|
2q35
|
| WNT7A |
Wnt family member 7A |
|
|
3p25.1
|
| WNT7B |
Wnt family member 7B |
|
|
22q13.31
|
| WNT8A |
Wnt family member 8A |
|
WNT8D |
5q31.2
|
| WNT8B |
Wnt family member 8B |
|
|
10q24.31
|
| WNT9A |
Wnt family member 9A |
WNT14 |
|
1q42.13
|
| WNT9B |
Wnt family member 9B |
WNT15 |
WNT14B |
17q21.32
|
| WNT10A |
Wnt family member 10A |
|
|
2q35
|
| WNT10B |
Wnt family member 10B |
|
"WNT-12, SHFM6" |
12q13.12
|
| WNT11 |
Wnt family member 11 |
|
|
11q13.5
|
| WNT16 |
Wnt family member 16 |
|
|
7q31.31
|
| Links: Developmental Signals - Wnt | OMIM Wnt1 | HGNC | Bmp Family | Fgf Family | Pax Family | R-spondin Family | Sox Family | Tbx Family | Wnt Family
|
|
|
|
Abnormality Genes
- triphalangeal thumb anomaly has been mapped to chromosome region 7q36 and caused by point mutations in the ZPA regulatory sequence (ZRS) which is a long-range cis-regulator for the SHH gene.[4][5]
- Monosomy 7q11.23, 7q11.23 deletion) Williams Syndrome (WS) is characterized by cardiovascular disease (elastin arteriopathy, peripheral pulmonary stenosis, supravalvular aortic stenosis, hypertension), distinctive facies, connective tissue abnormalities, mental retardation (usually mild), a specific cognitive profile, unique personality characteristics, growth abnormalities, and endocrine abnormalities (hypercalcemia, hypercalciuria, hypothyroidism, and early puberty).
External Links
External Links Notice - The dynamic nature of the internet may mean that some of these listed links may no longer function. If the link no longer works search the web with the link text or name. Links to any external commercial sites are provided for information purposes only and should never be considered an endorsement. UNSW Embryology is provided as an educational resource with no clinical information or commercial affiliation.
Cite this page: Hill, M.A. (2024, April 25) Embryology Genetics - Chromosome 7. Retrieved from https://embryology.med.unsw.edu.au/embryology/index.php/Genetics_-_Chromosome_7
- What Links Here?
- © Dr Mark Hill 2024, UNSW Embryology ISBN: 978 0 7334 2609 4 - UNSW CRICOS Provider Code No. 00098G
Cite this page: Hill, M.A. (2024, April 25) Embryology Genetics - Chromosome 7. Retrieved from https://embryology.med.unsw.edu.au/embryology/index.php/Genetics_-_Chromosome_7
- What Links Here?
- © Dr Mark Hill 2024, UNSW Embryology ISBN: 978 0 7334 2609 4 - UNSW CRICOS Provider Code No. 00098G
- ↑ Wlodarski MW, Sahoo SS & Niemeyer CM. (2018). Monosomy 7 in Pediatric Myelodysplastic Syndromes. Hematol. Oncol. Clin. North Am. , 32, 729-743. PMID: 30047423 DOI.
- ↑ Inaba T, Honda H & Matsui H. (2018). The enigma of monosomy 7. Blood , 131, 2891-2898. PMID: 29615405 DOI.
- ↑ Velker BAM, Denomme MM, Krafty RT & Mann MRW. (2017). Maintenance ofMestimprinted methylation in blastocyst-stage mouse embryos is less stable than other imprinted loci following superovulation or embryo culture. Environ Epigenet , 3, dvx015. PMID: 29492315 DOI.
- ↑ Sun M, Ma F, Zeng X, Liu Q, Zhao XL, Wu FX, Wu GP, Zhang ZF, Gu B, Zhao YF, Tian SH, Lin B, Kong XY, Zhang XL, Yang W, Lo WH & Zhang X. (2008). Triphalangeal thumb-polysyndactyly syndrome and syndactyly type IV are caused by genomic duplications involving the long range, limb-specific SHH enhancer. J. Med. Genet. , 45, 589-95. PMID: 18417549 DOI.
- ↑ Potuijt JWP, Baas M, Sukenik-Halevy R, Douben H, Nguyen P, Venter DJ, Gallagher R, Swagemakers SM, Hovius SER, van Nieuwenhoven CA, Galjaard RH, van der Spek PJ, Ahituv N & de Klein A. (2018). A point mutation in the pre-ZRS disrupts sonic hedgehog expression in the limb bud and results in triphalangeal thumb-polysyndactyly syndrome. Genet. Med. , , . PMID: 29543231 DOI.
{kind=link}



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
.jpg){kind=link}
.jpg){kind=link}
.jpg){kind=link}
.jpg){kind=link}
{kind=link}
{kind=link}
{kind=link}