Gastrointestinal Tract - Abnormalities: Difference between revisions
mNo edit summary |
mNo edit summary |
||
| (2 intermediate revisions by the same user not shown) | |||
| Line 13: | Line 13: | ||
==International Classification of Diseases== | ==International Classification of Diseases== | ||
{{ICD GIT table}} | {{ICD GIT table}} | ||
{{ICD-11 GIT table}} | |||
==Some Recent Findings== | ==Some Recent Findings== | ||
| Line 147: | Line 149: | ||
:'''Links:''' [http://www.ncbi.nlm.nih.gov/pmc/articles/PMC1493602/figure/F5 Image - small intestine duplication] | :'''Links:''' [http://www.ncbi.nlm.nih.gov/pmc/articles/PMC1493602/figure/F5 Image - small intestine duplication] | ||
==Oesophagus== | |||
The current [[International Classification of Diseases]] (ICD-10) code XVII Congenital Malformations [[International_Classification_of_Diseases_-_XVII_Congenital_Malformations#Q39_Congenital_malformations_of_oesophagus|'''Q39''' Congenital malformations of oesophagus]] | |||
* Q39.0 Atresia of oesophagus without fistula Atresia of oesophagus NOS | |||
* Q39.1 Atresia of oesophagus with tracheo-oesophageal fistula Atresia of oesophagus with broncho-oesophageal fistula | |||
* Q39.2 Congenital tracheo-oesophageal fistula without atresia Congenital tracheo-oesophageal fistula NOS | |||
* Q39.3 Congenital stenosis and stricture of oesophagus | |||
* Q39.4 Oesophageal web | |||
* Q39.5 Congenital dilatation of oesophagus | |||
* Q39.6 Diverticulum of oesophagus Oesophageal pouch | |||
* Q39.8 Other congenital malformations of oesophagus Absent Congenital displacement Duplication (of) oesophagus | |||
* Q39.9 Congenital malformation of oesophagus, unspecified | |||
Note ICD-10 is currently being updated to [[International_Classification_of_Diseases#International_Classification_of_Diseases_11_.28ICD11.29|ICD-11]] and will have new replacement coding. | |||
===Oesophageal Atresia with Tracheo-Oesophageal Fistula=== | |||
(Q39.1 Atresia of oesophagus with tracheo-oesophageal fistula Atresia of oesophagus with broncho-oesophageal fistula, OA/TOF) | |||
:[http://www.who.int/classifications/icd/revision/en/ ICD-11] (beta) [http://id.who.int/icd/entity/1582061097 '''LB12.2 Atresia of oesophagus'''] "Oesophageal atresia encompasses a group of congenital anomalies with an interruption in the continuity of the oesophagus, with or without persistent communication with the trachea. In 86% of cases there is a distal tracheooesophageal fistula, in 7% of cases there is no fistulous connection, while in 4% of cases there is a tracheooesophageal fistula without atresia. The remaining cases are made up of patients with OA with proximal, or both proximal and distal, tracheooesophageal fistula." | |||
This abnormality has been shown to be associated with Tbx1 mutations that also include DiGeorge syndrome.<ref name=PMID24356861><pubmed>24356861</pubmed></ref> | |||
:'''Links:''' [[Developmental Signals - Tbx|TBX]] | [[2016_Group_Project_5|Student Project - Di George syndrome]] | [https://www.omim.org/entry/602054 OMIM - TBX1] | [https://www.omim.org/entry/188400 OMIM - DiGeorge] | |||
== Intestinal Malrotation == | == Intestinal Malrotation == | ||
Revision as of 11:56, 20 July 2017
| Embryology - 23 Apr 2024 |
|---|
| Google Translate - select your language from the list shown below (this will open a new external page) |
|
العربية | català | 中文 | 中國傳統的 | français | Deutsche | עִברִית | हिंदी | bahasa Indonesia | italiano | 日本語 | 한국어 | မြန်မာ | Pilipino | Polskie | português | ਪੰਜਾਬੀ ਦੇ | Română | русский | Español | Swahili | Svensk | ไทย | Türkçe | اردو | ייִדיש | Tiếng Việt These external translations are automated and may not be accurate. (More? About Translations) |
Introduction
The "simple tube" of the gastrointestinal tract and its associated organs have many different tract and organ specific abnormalities. Due to the complex nature (different germ layer contributions, organogenisis) of the growth, elongation and folding of the tract, there are also several mechanical disorders of folding (rotation). Musculoskeletal abnormalities of the anterior body wall can also result in gastrointestinal abnormalities.
Note that as this system begins function (digestively) postnatally, unless there is a determined genetic history within the family, several abnormalities only become evident postnatally, in particular, metabolic disorders often identified by the Guthrie test.
International Classification of Diseases
| ICD10 Other congenital malformations of the digestive system (Q38-Q45) | ||||||
|---|---|---|---|---|---|---|
| XVII Congenital Malformations - Other congenital malformations of the digestive system (Q38-Q45) | ||||||
| Q38 Other congenital malformations of tongue, mouth and pharynx
Excl.: macrostomia (Q18.4) microstomia (Q18.5)
| ||||||
Q39 Congenital malformations of oesophagus
| ||||||
Q40 Other congenital malformations of upper alimentary tract
| ||||||
| Q41 Congenital absence, atresia and stenosis of small intestine
Incl.: congenital obstruction, occlusion and stricture of small intestine or intestine NOS Excl.: meconium ileus (E84.1)
| ||||||
| Q42 Congenital absence, atresia and stenosis of large intestine
Incl.: congenital obstruction, occlusion and stricture of large intestine
| ||||||
Q43 Other congenital malformations of intestine
| ||||||
Q44 Congenital malformations of gallbladder, bile ducts and liver
| ||||||
| Q45 Other congenital malformations of digestive system
Excl.: congenital: diaphragmatic hernia (Q79.0) hiatus hernia (Q40.1)
| ||||||
|
World Health Organisation. International Statistical Classification of Diseases and Related Health Problems. (1992) 10th Revision (ICD-10). Geneva: WHO ICD-10 - 2016 Online (English) | ||||||
Links: Gastrointestinal Abnormalities
| ||||||
| ICD10 - Gastrointestinal | Genital | Renal | Integumentary |
Some Recent Findings
|
| More recent papers |
|---|
 This table allows an automated computer search of the external PubMed database using the listed "Search term" text link.
More? References | Discussion Page | Journal Searches | 2019 References | 2020 References Search term: Abnormal Development Gastrointestinal Tract <pubmed limit=5>Abnormal Development Gastrointestinal Tract</pubmed> |
Movies
|
|
|
|

Statistics
Australian

|
The pie diagram shows the relative contribution of major gastrointestinal tract abnormalities as a percentage of the total number of congenital abnormalities in Australia beween 1981 - 92.
Note that the digestive system represents approximately 6% of all major congenital abnormalities. One of the most common abnormalities occurring in (2% - 3% population) is Meckel's Diverticulum. The mouth (cleft lip, cleft palate) is part of the digestive tract, but more accurately reflects an abnormality of face formation. |
| Data shown as a percentage of all major abnormalities based upon published statistics using the same groupings as Congenital Malformations Australia 1981-1992 P. Lancaster and E. Pedisich ISSN 1321-8352. |
| Australian GIT Abnormalities (2002-2003) |
|---|
Oesophageal atresia/stenosis - (2.0 per 10,000 births) ICD-10 Q39.0–Q39.3
|
Small intestinal atresia/stenosis - (2.4 per 10,000 births) ICD-10 Q41.0-Q41.2
|
Anorectal atresia/stenosis - ( 3.1 per 10,000 births) ICD-10 Q42.0–Q42.3
|
Hirschsprung’s disease - (1.3 per 10,000 births) ICD-10 Q43.1
|
Exomphalos - (Omphalocele) (2.1 per 10,000 births) ICD-10 Q79.2
|
Gastroschisis - (2.6 per 10,000 births) ICD-10 Q79.3
|
|
| Australian Palate Abnormalities (2002-2003) |
|---|
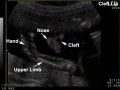
| Cleft lip with or without cleft palate (9.2 per 10,000 births) ICD-10 Q36.0, Q36.1, Q36.9, Q37.0–Q37.5, Q37.8, Q37.9 |
A congenital anomaly characterised by a partial or complete clefting of the upper lip, with or without clefting of the alveolar ridge or the hard palate. Excludes a midline cleft of the upper or lower lip and an oblique facial fissure (going towards the eye).
|
| Cleft palate without cleft lip (8.1 per 10,000 births) ICD-10 Q35.0–Q35.9 |
A congenital anomaly characterised by a closure defect of the hard and/or soft palate behind the foramen incisivum without a cleft lip. This anomaly includes sub-mucous cleft palate, but excludes cleft palate with a cleft lip, a functional short palate and high narrow palate.
|
|
USA Selected
CDC National estimates for selected GIT related major birth defects (2004–2006).
| Birth Defects | Cases per Births (1 in ...) | Estimated Annual Number of Cases |
|---|---|---|
| Cleft palate without cleft lip | 1,574 | 2,651 |
| Cleft lip with and without cleft palate | 940 | 4,437 |
| Esophageal atresia/tracheoesophageal fistula | 4,608 | 905 |
| Rectal and large intestinal atresia/stenosis | 2,138 | 1,952 |
| Gastroschisis | 2,229 | 1,871 |
| Omphalocele | 5,386 | 775 |
| Diaphragmatic hernia | 3,836 | 1,088 |
| Trisomy 21 (Down syndrome) | 691 | 6,037 |
Some Recent Findings
|
| More recent papers |
|---|
This table allows an automated computer search of the external PubMed database using the listed "Search term" text link.
More? References | Discussion Page | Journal Searches | 2019 References | 2020 References Search term: Abnormal Development Gastrointestinal Tract <pubmed limit=5>Abnormal Development Gastrointestinal Tract</pubmed> |
GIT Lumen Abnormalities

There are several types of abnormalities (atresia, stenosis and duplication) that impact upon the continuity of the gastrointestinal tract lumen.
Atresia
An interruption of the lumen (esophageal atresia, duodenal atresia, extrahepatic biliary atresia, anorectal atresia). See oesophageal Atresia review[6]
- Pyloric atresia (PA) - a very rare condition (incidence 1 in 100,000 newborns) and about 1% of all intestinal atresias.
| Oesophageal Atresia[7] | |
|---|---|

|

|
| Oesophageal atresia x-ray | Classification of Oesophageal atresia |
Stenosis
A narrowing of the lumen (esophageal stenosis, duodenal stenosis, pyloric stenosis)
- esophageal stenosis[8]
Duplication
An incomplete recanalization resulting in parallel lumens, this is really a specialized form of stenosis.
Oesophagus
The current International Classification of Diseases (ICD-10) code XVII Congenital Malformations Q39 Congenital malformations of oesophagus
- Q39.0 Atresia of oesophagus without fistula Atresia of oesophagus NOS
- Q39.1 Atresia of oesophagus with tracheo-oesophageal fistula Atresia of oesophagus with broncho-oesophageal fistula
- Q39.2 Congenital tracheo-oesophageal fistula without atresia Congenital tracheo-oesophageal fistula NOS
- Q39.3 Congenital stenosis and stricture of oesophagus
- Q39.4 Oesophageal web
- Q39.5 Congenital dilatation of oesophagus
- Q39.6 Diverticulum of oesophagus Oesophageal pouch
- Q39.8 Other congenital malformations of oesophagus Absent Congenital displacement Duplication (of) oesophagus
- Q39.9 Congenital malformation of oesophagus, unspecified
Note ICD-10 is currently being updated to ICD-11 and will have new replacement coding.
Oesophageal Atresia with Tracheo-Oesophageal Fistula
(Q39.1 Atresia of oesophagus with tracheo-oesophageal fistula Atresia of oesophagus with broncho-oesophageal fistula, OA/TOF)
- ICD-11 (beta) LB12.2 Atresia of oesophagus "Oesophageal atresia encompasses a group of congenital anomalies with an interruption in the continuity of the oesophagus, with or without persistent communication with the trachea. In 86% of cases there is a distal tracheooesophageal fistula, in 7% of cases there is no fistulous connection, while in 4% of cases there is a tracheooesophageal fistula without atresia. The remaining cases are made up of patients with OA with proximal, or both proximal and distal, tracheooesophageal fistula."
This abnormality has been shown to be associated with Tbx1 mutations that also include DiGeorge syndrome.[9]
Intestinal Malrotation
A recent study[10] has suggested that malrotation may result from the stunted embryonic development of intestinal secondary loops.

|
Presents clinically in symptomatic malrotation as:
Neonates - bilious vomiting and bloody stools. Newborn - bilious vomiting and failure to thrive. Infants - recurrent abdominal pain, intestinal obstruction, malabsorption/diarrhea, peritonitis/septic shock, solid food intolerance, common bile duct obstruction, abdominal distention, and failure to thrive. |

Ladd's Bands[11] A series of bands crossing the duodenum which can cause duodenal obstruction. |
Volvulus
Twisting of the midgut (bowel) which causes obstruction to the flow of material. Can include a variable loss of local blood supply which leads to tissue death.

|

|

|
Diagnosis is generally by upper gastrointestinal radiologic examination or less frequently by barium enema or CT scan.
Corrective surgery is generally by the Ladd's procedure, even with surgical treatment there is still significant associated complications and long-term morbidity.
What abnormal embryological processes could interfere with normal rotation and fixation of the gut?
Search PubMed: intestinal+malrotation
OMIM: Volvulus of Midgut
Links: Medlineplus - childhood volvulus | AAFP - Bilious Vomiting in the Newborn | Pediatric education - Neonatal Bilious Emesis |
Situs Inversus Viscera
Disturbance of the lateralisation of the liver may produce transposition of some or all of the foregut and its derivatives.
Also in situs inversus the anatomical relations of the duodenum, pancreas, bile ducts and portal veins may be reversed or disordered.
Search PubMed: Situs Inversus Viscera
Meckel's Diverticulum



This GIT abnormality is a very common (incidence of 1–2% in the general population) and results from improper closure and absorption of the omphalomesenteric duct (vitelline duct) in development. This transient developmental duct connects the yolk to the primitive gastrointestinal tract.
In addition to Meckel's diverticulum there are a range of other vitelline duct abnormalities, which depend on the degree from a completely patent duct at the umbilicus to lesser remnants (cysts, fibrous cords connecting umbilicus to distal ileum, granulation tissue at umbilicus, or umbilical hernias).
- Links: OMIM - Meckel's Diverticulum | Pubmed - Meckel's Diverticulum | Pubmed - omphalomesenteric duct | Pubmed - vitelline duct
Intestinal Aganglionosis
(intestinal aganglionosis, Hirschsprung's disease, aganglionic colon, megacolon, congenital aganglionic megacolon, congenital megacolon) A condition caused by the lack of enteric nervous system (neural ganglia) in the intestinal tract responsible for gastric motility (peristalsis). In general, its severity is dependent upon the amount of the GIT that lacks intrinsic ganglia, due to developmental lack of neural crest migration into those segments. (More? Neural Crest System - Abnormalities)
Historically, Hirschsprung's disease takes its name from Dr Harald Hirschsprung (1830-1916) a Danish pediatrician (of German extraction). In 1886, he presented at the German Society of Pediatrics conference in Berlin a case of 2 infants who died of complications of bowel obstruction (H. Hirschsprung, Stuhltragheit Neugeborener in Folge von Dilatation und Hypertrophie des Colons, Jhrb f Kinderh 27 (1888), pp. 1-7). Later autopsies identified a dilatation and hypertrophy of large intestine, and the rectum appeared normally narrow. Hirschsprung suggested that the condition was an inborn disease and named it congenital megacolon.
The first indication in newborns is an absence of the first bowel movement, other symptoms include throwing up and intestinal infections. Clinically this is detected by one or more tests (barium enema and x ray, manometry or biopsy) and can currently only be treated by surgery. A temoporary ostomy (Colostomy or Ileostomy) with a stoma is carried out prior to a more permanent pull-through surgery.

|

|
|
| Ostomy - Aganglionic portion removed | Stoma - intestine attached to the abdomen wall | |

|

|

|
| Short section of the colon without smooth muscle neural ganglia | Aganglionic segment removed | Reattachment |
Images: NIH - NIDDK - Hirschsprungs
- Links: Enteric Nervous System | Neural Crest System - Abnormalities | NIH - NIDDK - Hirschsprungs | MedlinePlus - Hirschsprung's disease | OMIM 142623 | Search PubMed
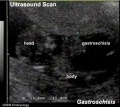
Gastroschisis
By definition, it is a body wall musculoskeletal defect, not a gastrointestinal tract defect, which in turn impacts upon GIT development.
| Gastroschisis (paraomphalocele, laparoschisis, abdominoschisis, abdominal hernia) is an abdominal wall defect associated with evisceration of the intestine (2.5 cases/10,000 births), occuring more frequently in young mothers (less than 20 years old). There have been reports that this abnormality is increasing in incidence.
The abnormality is usually situated to the right of the umbilicus and abdominal contents, mainly gastrointestinal, are found outside the anterior body wall. International Classification of Diseases, 9th Revision diagnosis (756.79) and procedure (54.71) code for gastroschisis (2003 to 2008). |

| |||
| Can occur in isolation and also in association with other gastrointestinal anomalies (intestinal atresia, perforation, necrosis or volvulus). Defects in other organ systems have been reported in up to 35% of children. There are several theories as to the cause of this abdominal wall defect, including recently failure of the yolk sac and related vitelline structures to be incorporated into the umbilical stalk.[12]
The condition can often be detected by ultrasound scan. Reported as a high mortality defect, only 60% of children survive until the end of the first year of age. |
| |||
Gastroschisis patients are commonly small for gestational age (SGA, birth weight < 10th centile). Frequency line graphs of the birth weight distribution, Kolmogorov-Smirnov test confirmed that this difference was significant (p < 0.001).[13]
|

|
Gastroschisis Classification
There has been a recent attempt to classify gastroschisis in order to measure clinical outcomes.[14]
- Simple gastroschisis - intact continuous bowel that is not compromised or breached at delivery or presentation
- Complex gastroschisis - presence of one or more of intestinal atresia, perforation or intestinal necrosis at delivery or presentation, or missed atresia
- Links: Gastroschisis
Omphalocele


An abnormality appearing similar to gastroschisis, involves "covered by membranes" and a lack of normal return of the bowel to the abdominal cavity and has a different position relative to the umbilical cord. The origin differs, as this is a failure of midgut loops to return to the body cavity after initial herniation into the umbilical cord during week 6 - 10.
- omphalocele - herniation of the bowel, liver and other organs into the intact umbilical cord, the tissues being covered by membranes unless the latter are ruptured
- gastroschisis - intact umbilical cord and evisceration of the bowel through a defect in the abdominal wall, generally to the right of the cord, with no membrane covering
Short-Bowel Syndrome
Not generally a developmental abnormality, but related to therapeutic intervention in GIT abnormalities or disease.
Short bowel syndrome is a group of problems affecting people who have had half or more of their small intestine removed. The most common reason for removing part of the small intestine is to treat Crohn's disease. Short bowel syndrome is treated through changes in diet, intravenous feeding, vitamin and mineral supplements, and medicine to relieve symptoms. (NDDIC)
- Links: Better Health Short Bowel syndrome | National Digestive Diseases Information Clearinghouse Short Bowel syndrome
Obstetric Cholestasis
A recent paper in the British Medical Journal discusses this pregnancy associated disease.
"Obstetric cholestasis (or intrahepatic cholestasis of pregnancy) remains widely disregarded as an important clinical problem, with many obstetricians still considering its main symptom, pruritus, a natural association of pregnancy. Obstetric cholestasis is associated with cholesterol gallstones. It may be extremely stressful for the mother but also carries risks for the baby." Piotr Milkiewicz, Elwyn Elias, Catherine Williamson, and Judith Weaver BMJ 2002; 324: 123-124
Small Bowel Obstruction
The are two major forms of small bowel obstruction are from either external (extrinsic) or internal (intrinsic) causes. Listed below are a few examples of both causes.
- Extrinsic Causes - adhesions, closed loop, strangulation, hernia and extrinsic masses
- Intrinsic Causes - intestinal malrotation, Crohn disease, adenocarcinoma, tuberculosis, radiation enteropathy, intramural hemorrhage, intussusception and intraluminal causes.
(More? see PMID 11353110)
Necrotizing Enterocolitis
Necrotizing enterocolitis (NE) is the death of intestinal tissue that occurs postnatally in mainly in premature and low birth weight infants (1 in 2,000 - 4,000 births). The underdeveloped gastointestinal tract appears to be susceptible to bacteria, normally found within the tract,to spread widely to other regions where they damage the tract wall and may enter the bloodstream.
Those with a higher risk for this condition include:
- Premature infants
- Infants who are fed concentrated formulas
- Infants in a nursery where an outbreak has occurred
- Infants who have received blood exchange transfusions
Meconium Plug Syndrome
(functional immaturity of the colon) Term used to describe a transient disorder of the newborn colon, which is characterized by delayed passage of meconium (more than 24 to 48 h), intestinal dilatation and yellow/green vomiting. More common in premature infants and can be determined by radiological dye study.
A recent study[17] by looked at thecorrelation of meconium plug as identified radiologically covering 1994 to 2007, of 77 patients (mean gestational age 37.4 weeks, birth weight, 2977 g) Hirschsprung's disease was found in 10 patients (13%). "Although all patients with plugs and persistent abnormal stooling patterns should prompt a rectal biopsy and genetic probe, the incidence of Hirschsprung's and cystic fibrosis may not be as high as previously reported."
Appendix Duplication
Appendix duplication is an extremely rare congenital anomaly (0.004% to 0.009% of appendectomy specimens) first classified according to their anatomic location by Cave in 1936[18] and a later modified by Wallbridge in 1963[19], subsequently two more types of appendix abnormalities have been identified.[20][21]
Modified Cave-Wallbridge Classification (table from[22])
| Classification of types of appendix duplication |
Features |
| A | Single cecum with various degrees of incomplete duplication |
| B1 (bird type) | Two appendixes symmetrically placed on either side of the ileocecal valve |
| B2 (tenia coli type) | ne appendix arises from the cecum at the usual site, and the second
appendix branches from the cecum along the lines of the tenia at various distances from the first |
| B3 | One appendix arises from the usual site, and the second appendix arises from
the hepatic flexura |
| B4 | One appendix arises from the usual site, and the second appendix arises from
the splenic flexura |
| C | Double cecum, each with an appendix |
| Horseshoe appendix | One appendix has two openings into a common cecum |
| Triple appendix | One appendix arises from the cecum at the usual site, and two additional appendixes arise from the colon |
Anorectal Malformations

(ARMs) A group of many different abnormalities that can involve the distal anus and rectum as well as the urinary and genital tracts, for review see[23]. Occurring with an incidence of approximately 1 in 5000 live births.
|
Classification of non-syndromic anorectal malformations (ARM) | |
| Males |
Recto-perineal fistula |
| Recto-urethral-bulbar fistula | |
| Recto-urethral-prostatic fistula | |
| Recto-bladderneck fistula | |
| Imperforated anus without fistula | |
| Complex and unusual defects | |
|
| |
| Females |
Recto-perineal fistula |
| Recto-vestibular fistula | |
| Cloaca with short common channel (< 3 cm) | |
| Cloaca with long common channel (> 3 cm) | |
| Imperforated anus without fistula | |
|
| |
| Complex and unusual defects |
Cloacal extrophy, covered cloacal extra |
| Posterior cloaca | |
| Associated to presacral mass | |
| Rectal atresia | |
|
| |
|
Table Levitt and Peña (2007)[23] | |
Anal Atresia
Anal atresia or imperforate anus is an abnormality of incomplete anorectal region development occurring in about 1 in 5,000 infants. Resulting in accumulation of stool within the colon.
- rectum may end and not connect with the anus
- rectum may connect anatomically to the wrong region (urethra, bladder, or vagina).
- anus may be very narrowed or missing altogether.
Congenital Cloaca
Anal muscles and vagina wall do not form leading to a variable opening composing all or some of the rectum, vagina and bladder. Surgically requires a colostomy and other procedures to transfers a muscle from another part of the body to create a functioning sphincter at the anus.
References
- ↑ <pubmed>25676186</pubmed>
- ↑ <pubmed>24267509</pubmed>| Orphanet J Rare Dis.
- ↑ <pubmed>23568430</pubmed>
- ↑ <pubmed>21841314</pubmed>
- ↑ <pubmed>718292</pubmed>
- ↑ <pubmed>17498283</pubmed>
- ↑ <pubmed>22851858</pubmed>| PMC3406418 | World J Gastroenterol.
- ↑ <pubmed>24289834</pubmed>
- ↑ <pubmed>24356861</pubmed>
- ↑ <pubmed>26297675</pubmed>
- ↑ <pubmed>24761085</pubmed>| J Minim Access Surg.
- ↑ <pubmed>19419415</pubmed>
- ↑ <pubmed> 22004141 </pubmed>| BMC Pediatr.
- ↑ <pubmed>11150437</pubmed>
- ↑ <pubmed>22325297</pubmed>| PMC3295733 | Ann Surg Innov Res.
- ↑ <pubmed>21283718</pubmed>| PMC3024424 | PLoS One.
- ↑ <pubmed>18485962</pubmed>
- ↑ <pubmed>17104589</pubmed>
- ↑ <pubmed>13998581</pubmed>
- ↑ <pubmed>2772830</pubmed>
- ↑ <pubmed>5635427</pubmed>
- ↑ <pubmed>21513538</pubmed>| J Medical Case Reports | PDF
- ↑ 23.0 23.1 23.2 <pubmed>17651510</pubmed>
Reviews
<pubmed>20549505</pubmed>| PMC2908440 <pubmed>15378215</pubmed> <pubmed>15026601</pubmed> <pubmed>12738470</pubmed> <pubmed>11826631</pubmed> <pubmed>11353110</pubmed>
Articles
<pubmed>19419414</pubmed> <pubmed>17230493</pubmed> <pubmed>16465538</pubmed> <pubmed>16390394</pubmed> <pubmed>16205928</pubmed> <pubmed>16563666</pubmed> <pubmed>15793730</pubmed> <pubmed>15488122</pubmed> <pubmed>12557047</pubmed>
Search PubMed
Search Pubmed: gastrointestinal tract abnormalities | intestinal malrotation | Situs Inversus Viscera | Gastroschisis | Intestinal Aganglionosis
External Links
External Links Notice - The dynamic nature of the internet may mean that some of these listed links may no longer function. If the link no longer works search the web with the link text or name. Links to any external commercial sites are provided for information purposes only and should never be considered an endorsement. UNSW Embryology is provided as an educational resource with no clinical information or commercial affiliation.
- NIH National Institute of Diabetes and Digestive and Kidney Diseases Hirschsprung Disease | Anatomic Problems of the Colon
- BMC Gastroenterology
Glossary Links
- Glossary: A | B | C | D | E | F | G | H | I | J | K | L | M | N | O | P | Q | R | S | T | U | V | W | X | Y | Z | Numbers | Symbols | Term Link
Cite this page: Hill, M.A. (2024, April 23) Embryology Gastrointestinal Tract - Abnormalities. Retrieved from https://embryology.med.unsw.edu.au/embryology/index.php/Gastrointestinal_Tract_-_Abnormalities
- © Dr Mark Hill 2024, UNSW Embryology ISBN: 978 0 7334 2609 4 - UNSW CRICOS Provider Code No. 00098G