ANAT2341 Lab 3 2013: Difference between revisions
No edit summary |
No edit summary |
||
| (6 intermediate revisions by the same user not shown) | |||
| Line 27: | Line 27: | ||
Standard methods can only identify large scale changes and rearrangements. However, since the development of Fluoresecence In-Situ Hybridisation (FISH) it is now possible to identify small scale changes such as microdeletions (e.g. Williams-Beuren Syndrome). | Standard methods can only identify large scale changes and rearrangements. However, since the development of Fluoresecence In-Situ Hybridisation (FISH) it is now possible to identify small scale changes such as microdeletions (e.g. Williams-Beuren Syndrome). | ||
[http://www.chd-uk.co.uk/types-of-chd-and-operations/williams-syndrome/ Williams syndrome page including FISH image] | |||
FISH can be used to detect the Philadelphia Chromosome also [http://www.cambridgebluegnome.com/applications/copy-number-imbalance/traditional-techniques/ BCR/ABL Fusion] | |||
==Human Chromosomes== | |||
[[File:Human_idiogram.jpg|thumb|300px|Human idiogram]] | |||
{{Human chromosomes}} | |||
==Some Human Disease Gene Locations== | |||
[[File:Human genetics chromosomes 1-4.jpg]] | |||
[[File:Human genetics chromosomes 5-8.jpg]] | |||
[[File:Human_genetics_chromosomes_9-12.jpg]] | |||
[[File:Human genetics chromosomes 17-20.jpg]] | |||
[[File:Human genetics chromosomes 21-XY.jpg]] | |||
==Genetic Inheritance== | |||
The figures below show the pattern of inheritance of a range of genetic disorders. In addition to these patterns are the known effects of increased maternal age and the effects of genetic mutations in the embryo and newborn. | |||
<gallery> | |||
File:Autosomal_dominant_inheritance.jpg|Autosomal dominant inheritance | |||
File:Autosomal recessive inheritance.jpg|Autosomal recessive inheritance | |||
File:X-Linked_dominant_(affected_father).jpg|X-Linked dominant (affected father) | |||
File:X-Linked_dominant_(affected_mother).jpg|X-Linked dominant (affected mother) | |||
File:X-Linked_recessive_(affected_father).jpg|X-Linked recessive (affected father) | |||
File:X-Linked recessive (carrier mother).jpg|X-Linked recessive (carrier mother) | |||
File:Mitochondrial_inheritance.jpg|Mitochondrial genome inheritance | |||
File:Codominant inheritance.jpg|Codominant inheritance | |||
</gallery> | |||
{{Template:GHR Inheritance}} | |||
An example of autosomal recessive traits are the majority of 50 or so lysosomal diseases, with two X-linked exceptions, that of Fabry disease and Mucopolysaccharidosis II.<ref name=PMID2050994><pubmed>2050994</pubmed>| [http://www.ojrd.com/content/5/1/14 Orphanet J Rare Dis.]</ref> | |||
===Trisomy=== | |||
In humans, the most common trisomy is trisomy 21 or Down syndrome. | |||
The term trisomy refers to the abnormal copy number of a specific chromosome, that is 3 copies instead of 2. The abnormality is identified by the chromosome that is present as 3 copies within the cell. | |||
* [[Trisomy 21]] | |||
* [[Trisomy 18]] | |||
* [[Trisomy 13]] | |||
* [[Trisomy X]] | |||
===Linkage analysis=== | |||
Identification of genetic mutations causing a disease are sometimes unknown and a common strategy to map the location of the mutation is to use linkage analysis. | |||
This strategy relies on establishing a family group with some affected by the disease and other that are unaffected. The larger the family, or the greater the number of different families with the same disease, the greater the power of the mapping approach. This technique relies on the knowledge that chromosomes recombine during meiosis. Small regions of chromosomes will generally be inherited as a series of single intact pieces. However, the larger the region, the greater the likelihood that it will be divided by meiotic recombination. By looking for correlation between segregation of the disease and co-segregation of genetic markers distributed all over the human genome, it is possible to identify chromosomal sites where specific markers are always (or nearly always) inherited when the disease is present. Once the specific chromosomal region has been mapped, genes in the vicinity must be identified and candidate genes that look likely to be causative can be sequenced to look for specific changes that might explain the defect. | |||
[http://www.ncbi.nlm.nih.gov/books/NBK7560/ Genetic mapping of Mendelian characters] | |||
==Modern methods in human genetics== | ==Modern methods in human genetics== | ||
===Genome-wide association study (GWAS)=== | |||
The National Human Genome Research Institute (USA) lists publications as part of the genome-wide association study (GWAS) in an attempt to assay at least 100,000 single nucleotide polymorphisms (SNPs) in the initial stage. | |||
:'''Links:''' [http://www.genome.gov/gwastudies/ Genome-wide association study] | [http://www.genome.gov National Human Genome Research Institute] | |||
GWAS is a cutting-edge form of linkage analysis that uses very high numbers of markers and large sample groups of the human population to look for genes that predispose individuals towards specific common diseases such as coronary artery disease or type I diabetes. The first successful GWAS study was published in 2005 on age-related macular degeneration. [http://www.ncbi.nlm.nih.gov/pubmed/15761122 Complement factor H polymorphism in age-related macular degeneration] | |||
===Copy Number Variations (CNV)=== | |||
Large deletions or duplications can be detected by cytogenetics or FISH analysis described above but smaller changes in copy number are difficult to detect. | |||
Some modern approaches include the use of real-time PCR assays that are capable of quantifying the number of copies of a gene in the genome. However, this strategy requires the use of individual PCR primers and can be slow and expensive. | |||
A modern technique that is capable of detecting very small CNVs in a multiplex assay is the Multiplex Ligation-dependent Probe Amplification (MLPA) assay. This system is a cost-effective way of measuring the dosage of up to 50 different probes in a gene region in a single PCR assay. This allows the detection of small mutations in candidate genes without prior knowledge of what or where the mutation might be. Assay kits are available for single gene mutations such as the breast cancer susceptibility genes BRCA1 and BRCA2 and larger deletions such as DiGeorge Syndrome and Prader-Willi/Angelman Syndromes. | |||
===Point Mutations=== | |||
Changes to single DNA bases can create nonsense (introduction of unexpected stop codons) or missense mutations (alteration of amino acid) or affect gene splicing. These mutations are the hardest to detect unless the likely cause is already known in advance because of the common frequency of such mutations in the human population. In this case, these mutations can be detected by PCR or PCR-based variations such as PCR RFLP, Allele-specific oligonucleotide hybridisation (ASO), Allele-specific PCR amplification refractory mutation system (ARMS) or Real-time PCR and melt-curve analysis. | |||
===Next generation sequencing=== | |||
Increasingly, there is a desire to bring the sequencing of the entire genome within reach of standard diagnostic costs. The speed and efficiency of these systems has undergone dramatic improvements over the last few years. The concept of the "$1000 genome", which means the point at which the cost per complete genome sequence drops to US$1000, describes a point at which a new era in personalized medicine will become affordable. | |||
However, it must be remembered that sequence data alone is not sufficient to identify causative mutations. Base changes are very common in the genome and are referred to as Single Nucleotide Polymorphisms (SNPs). Most of these SNPs are functionally neutral. Determining the one base change that causes the disease amongst these millions of SNPs is a hard task and may require linkage data to identify the causative gene. If the base change causes obvious nonsense or missense changes to the identified gene then this is a strong indicator. However, some base changes can cause more subtle changes in splicing or gene expression and these will require extensive further research to identify. | |||
{{Glossary}} | {{Glossary}} | ||
{{Footer}} | {{Footer}} | ||
Latest revision as of 09:39, 21 August 2013
Human genetic diseases
This practical contains the first of a series of tutorials designed to support the group projects. We will discuss various techniques used to diagnose and identify the genetic abnormalities found in many human congential illnesses.
Objectives of this laboratory
- second short answer/ multiple choice test of last week's lecture material (Week 3 of human development)
- Tutorial on human genetic disorders, diagnosis, mapping and modern techniques such as genome sequencing.
- Provide time for groupwork and allow groups to ask questions of lecturing staff.
Human developmental diseases
There are many common and rare human genetic diseases. Some are due to mutations of a single gene - monogenic disorders e.g. Cystic Fibrosis, Duchenne Muscular Dystrophy Some are due to chromosomal abnormalities - polygenic disorders e.g. Williams-Beuren syndrome, Downs Syndrome
- (Some common genetic diseases) examples of specific genetic disorders NHGRI
- (Comprehensive database of human genetic disorders and associated genes) Online Mendelian Inheritance in Man OMIM
Diagnosis of human genetic diseases
- Clinical examination and assessment
- Personal history
- Family history
- Genetic tests
Genetic tests may include clinical cytogenetics which involves an examination of the chromosomes to look for evidence of aneuploidy, which means a loss or gain of the usual amount of genetic material, such as Trisomy 21 - Downs Syndrome. This will lead to changes in the number of copies of genes in the genome - a copy number variation or CNV.
This method will also pick up chromosomal rearrangements such as inversions or translocations. The breakpoints of these chromosomal rearrangements can lead to abnormal expression or complete loss of gene function as well as in some cases, creation of an abnormal chimeric fusion gene such as the Philadelphia Chromosome which causes a fusion of the genes BCR and ABL. The resulting protein leads to the development of chronic myelogenous leukaemia.
Standard methods can only identify large scale changes and rearrangements. However, since the development of Fluoresecence In-Situ Hybridisation (FISH) it is now possible to identify small scale changes such as microdeletions (e.g. Williams-Beuren Syndrome). Williams syndrome page including FISH image
FISH can be used to detect the Philadelphia Chromosome also BCR/ABL Fusion
Human Chromosomes

| |
| Idiogram Chromosome Banding - The term refers to the light and dark pattern, seen after staining with a dye, of individual chromosomes identified in metaphase. It is only in meiosis and mitosis during metaphase that chromosomes can be easily identified, during the normal cell life (interphase) the chromosomes are unravelled and distributed within the nucleus in chromosome territories. A band is that part of a chromosome which is clearly distinguishable from nearby regions by appearing darker or brighter with one or more banding techniques. | |
| Genetic abnormality locations: 1-4 | 5-8 | 9-12 | 13-16 | 17-20 | 21-XY | sSMC | |
| |
| Links: Genetics | Abnormal Development - Genetic |
Cite this page: Hill, M.A. (2024, April 24) Embryology ANAT2341 Lab 3 2013. Retrieved from https://embryology.med.unsw.edu.au/embryology/index.php/ANAT2341_Lab_3_2013
- © Dr Mark Hill 2024, UNSW Embryology ISBN: 978 0 7334 2609 4 - UNSW CRICOS Provider Code No. 00098G
Some Human Disease Gene Locations





Genetic Inheritance
The figures below show the pattern of inheritance of a range of genetic disorders. In addition to these patterns are the known effects of increased maternal age and the effects of genetic mutations in the embryo and newborn.
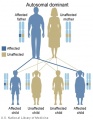
Autosomal dominant inheritance
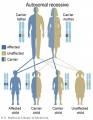
Autosomal recessive inheritance
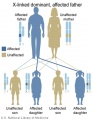
X-Linked dominant (affected father)
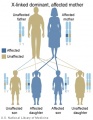
X-Linked dominant (affected mother)
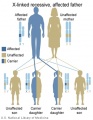
X-Linked recessive (affected father)
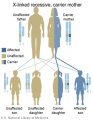
X-Linked recessive (carrier mother)
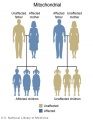
Mitochondrial genome inheritance
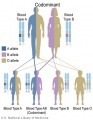
Codominant inheritance


.jpg)
.jpg)
.jpg)
.jpg)


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
- Inheritance Pattern images: Genetic Abnormalities | autosomal dominant | autosomal recessive | X-linked dominant (affected father) | X-Linked dominant (affected mother) | X-Linked recessive (affected father) | X-Linked recessive (carrier mother) | mitochondrial inheritance | Codominant inheritance | Genogram symbols | Genetics
An example of autosomal recessive traits are the majority of 50 or so lysosomal diseases, with two X-linked exceptions, that of Fabry disease and Mucopolysaccharidosis II.[1]
Trisomy
In humans, the most common trisomy is trisomy 21 or Down syndrome.
The term trisomy refers to the abnormal copy number of a specific chromosome, that is 3 copies instead of 2. The abnormality is identified by the chromosome that is present as 3 copies within the cell.
Linkage analysis
Identification of genetic mutations causing a disease are sometimes unknown and a common strategy to map the location of the mutation is to use linkage analysis. This strategy relies on establishing a family group with some affected by the disease and other that are unaffected. The larger the family, or the greater the number of different families with the same disease, the greater the power of the mapping approach. This technique relies on the knowledge that chromosomes recombine during meiosis. Small regions of chromosomes will generally be inherited as a series of single intact pieces. However, the larger the region, the greater the likelihood that it will be divided by meiotic recombination. By looking for correlation between segregation of the disease and co-segregation of genetic markers distributed all over the human genome, it is possible to identify chromosomal sites where specific markers are always (or nearly always) inherited when the disease is present. Once the specific chromosomal region has been mapped, genes in the vicinity must be identified and candidate genes that look likely to be causative can be sequenced to look for specific changes that might explain the defect.
Genetic mapping of Mendelian characters
Modern methods in human genetics
Genome-wide association study (GWAS)
The National Human Genome Research Institute (USA) lists publications as part of the genome-wide association study (GWAS) in an attempt to assay at least 100,000 single nucleotide polymorphisms (SNPs) in the initial stage.
GWAS is a cutting-edge form of linkage analysis that uses very high numbers of markers and large sample groups of the human population to look for genes that predispose individuals towards specific common diseases such as coronary artery disease or type I diabetes. The first successful GWAS study was published in 2005 on age-related macular degeneration. Complement factor H polymorphism in age-related macular degeneration
Copy Number Variations (CNV)
Large deletions or duplications can be detected by cytogenetics or FISH analysis described above but smaller changes in copy number are difficult to detect.
Some modern approaches include the use of real-time PCR assays that are capable of quantifying the number of copies of a gene in the genome. However, this strategy requires the use of individual PCR primers and can be slow and expensive.
A modern technique that is capable of detecting very small CNVs in a multiplex assay is the Multiplex Ligation-dependent Probe Amplification (MLPA) assay. This system is a cost-effective way of measuring the dosage of up to 50 different probes in a gene region in a single PCR assay. This allows the detection of small mutations in candidate genes without prior knowledge of what or where the mutation might be. Assay kits are available for single gene mutations such as the breast cancer susceptibility genes BRCA1 and BRCA2 and larger deletions such as DiGeorge Syndrome and Prader-Willi/Angelman Syndromes.
Point Mutations
Changes to single DNA bases can create nonsense (introduction of unexpected stop codons) or missense mutations (alteration of amino acid) or affect gene splicing. These mutations are the hardest to detect unless the likely cause is already known in advance because of the common frequency of such mutations in the human population. In this case, these mutations can be detected by PCR or PCR-based variations such as PCR RFLP, Allele-specific oligonucleotide hybridisation (ASO), Allele-specific PCR amplification refractory mutation system (ARMS) or Real-time PCR and melt-curve analysis.
Next generation sequencing
Increasingly, there is a desire to bring the sequencing of the entire genome within reach of standard diagnostic costs. The speed and efficiency of these systems has undergone dramatic improvements over the last few years. The concept of the "$1000 genome", which means the point at which the cost per complete genome sequence drops to US$1000, describes a point at which a new era in personalized medicine will become affordable.
However, it must be remembered that sequence data alone is not sufficient to identify causative mutations. Base changes are very common in the genome and are referred to as Single Nucleotide Polymorphisms (SNPs). Most of these SNPs are functionally neutral. Determining the one base change that causes the disease amongst these millions of SNPs is a hard task and may require linkage data to identify the causative gene. If the base change causes obvious nonsense or missense changes to the identified gene then this is a strong indicator. However, some base changes can cause more subtle changes in splicing or gene expression and these will require extensive further research to identify.
Glossary Links
- Glossary: A | B | C | D | E | F | G | H | I | J | K | L | M | N | O | P | Q | R | S | T | U | V | W | X | Y | Z | Numbers | Symbols | Term Link
Cite this page: Hill, M.A. (2024, April 24) Embryology ANAT2341 Lab 3 2013. Retrieved from https://embryology.med.unsw.edu.au/embryology/index.php/ANAT2341_Lab_3_2013
- © Dr Mark Hill 2024, UNSW Embryology ISBN: 978 0 7334 2609 4 - UNSW CRICOS Provider Code No. 00098G
- ↑ <pubmed>2050994</pubmed>| Orphanet J Rare Dis.